Removal of human reads from SARS-CoV-2 sequencing data
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How can you remove traces of human sequence data from a sequenced viral sample?
Requirements:
Obtain viral (SARS-CoV-2) sequencing data with contaminating human reads from public sources
Organize the data into a collection
Preprocess and map the data against the human reference genome
Eliminate reads/read pairs that map to the human genome from the original data
- Introduction to Galaxy Analyses
- slides Slides: Quality Control
- tutorial Hands-on: Quality Control
- slides Slides: Mapping
- tutorial Hands-on: Mapping
- tutorial Hands-on: Using dataset collections
Time estimation: 1 hourLevel: Intermediate IntermediateSupporting Materials:Published: Aug 4, 2021Last modification: Nov 9, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00236rating Rating: 5.0 (0 recent ratings, 7 all time)version Revision: 5
Patient samples for pathogen detection are usually “contaminated” with human host DNA. Such contamination, if not removed from sequencing data, may pose an issue with certain types of data analyses. Another issue is that submitting the raw sequenced reads with the human sequence “contamination” included to a public database may simply violate national or institutional regulations for handling patient data. For this second reason in particular, researchers will regularly have to remove traces of human sequencing data even from samples that, by their nature, should be highly enriched for just pathogen reads, as is the case, for example, for ampliconic viral samples, in which viral sequences have been PCR-amplified with virus-specific primers before sequencing.
This tutorial will guide you through a typical workflow for clearing human sequences from any kind (ampliconic or not) of viral sequenced sample, which retains non-human reads in a format ready to be submitted to public databases.
Comment: Nature of the input dataWe will use sequencing data of bronchoalveolar lavage fluid (BALF) samples obtained from early COVID-19 patients in China as our input data. Since such samples are expected to be contaminated signficantly with human sequenced reads, the effect of the cleaning steps will be much more apparent than for ampliconic sequencing data from diagnostic patient swabs. Nevertheless the cleaning processs does not rely on any particular sample isolation or preprcocessing method and could be used unaltered on, for example, ARTIC amplified SARS-CoV-2 samples.
AgendaIn this tutorial, we will deal with:
Get data
As always, it is best to give each analysis you are performing with Galaxy its own history, so let’s start with preparing this:
Hands On: Prepare new history
Create a new history for this tutorial and give it a proper name
To create a new history simply click the new-history icon at the top of the history panel:
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter

Getting the sequencing data
The sequenced reads datasets used in this tutorial (4 files representing 2 Illumina paired-end sequenced samples) have been deposited on Zenodo and can be uploaded to Galaxy via their URLs. After that, we will arrange the uploaded data into a collection in our Galaxy history to facilitate its handling in the analysis.
Hands On: Data upload and rearrangement
Upload the data from Zenodo via URLs
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
The URLs for our example data are these:
https://zenodo.org/record/3732359/files/SRR10903401_r1.fq.gz https://zenodo.org/record/3732359/files/SRR10903401_r2.fq.gz https://zenodo.org/record/3732359/files/SRR10903402_r1.fq.gz https://zenodo.org/record/3732359/files/SRR10903402_r2.fq.gzArrange the data into a paired dataset collection
- Click on galaxy-selector Select Items at the top of the history panel
- Check all the datasets in your history you would like to include
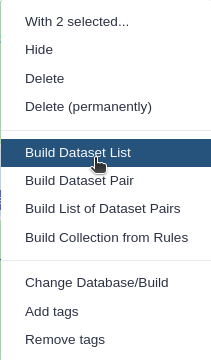
Click n of N selected and choose Advanced Build List
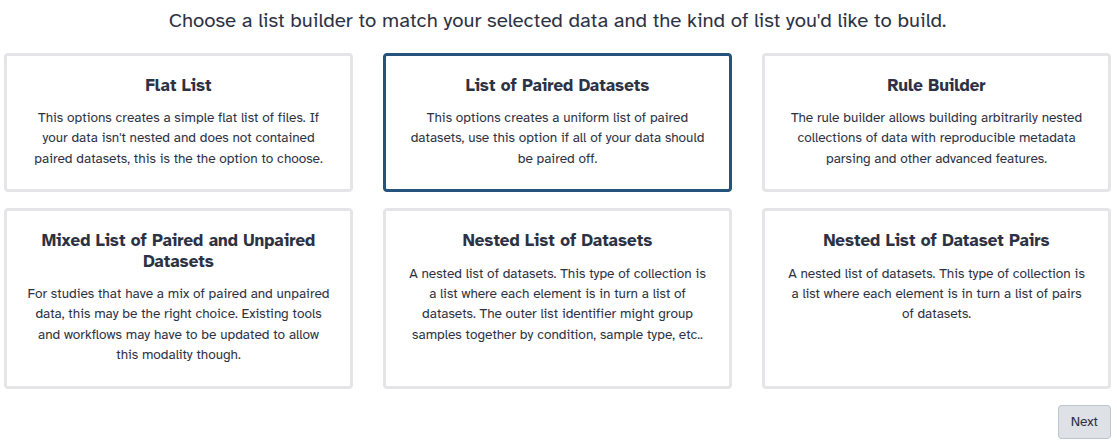
You are in the collection building wizard. Choose List of Paired Datasets and click ‘Next’ button at the right bottom corner.
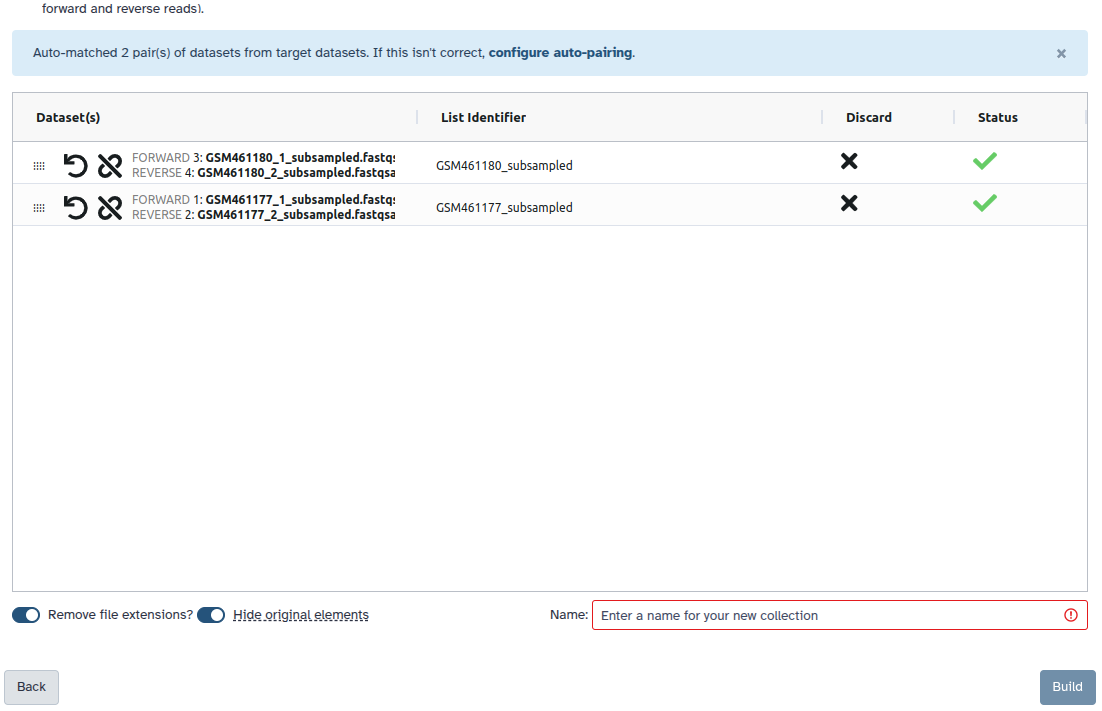
Check and configure auto-pairing. Commonly matepairs have suffix
_1and_2or_R1and_R2. Click on ‘Next’ at the bottom.
- Edit the List Identifier as required.
- Enter a name for your collection
- Click Build to build your collection
- Click on the checkmark icon at the top of your history again
For the example datasets this means:
- You need to tell Galaxy about the suffix for your forward and reverse reads, respectively:
- set the text of unpaired forward to:
_r1.fq.gz- set the text of unpaired reverse to:
_r2.fq.gz- click:
Auto-pairAll datasets should now be moved to the paired section of the dialog, and the middle column there should show that only the sample accession numbers, i.e.
SRR10903401andSRR10903402, will be used as the pair names.- Make sure Hide original elements is checked to obtain a cleaned-up history after building the collection.
- Click Create Collection

Read trimming and mapping
In order to learn which of the reads of each of the input samples are of likely human origin, we need to map the reads to the human reference genome. To improve the quality of this step we will also trim low-quality bases from the ends of all reads before passing them to the read mapping software.
CommentDo you want to learn more about the principles behind quality control (including trimming) and mapping? Follow our dedicated tutorials:
Hands On: Trim reads and map them to the human reference genome
- Trimmomatic ( Galaxy version 0.38.0) with default settings and:
- “Single-end or paired-end reads?”:
Paired-end (as collection)
- “Select FASTQ dataset collection with R1/R2 pair”: the collection of sequenced reads input datasets
- Map with BWA-MEM ( Galaxy version 0.7.17.2) with the following parameters:
- “Will you select a reference genome from your history or use a built-in index?”:
Use a built-in genome index
“Using reference genome”:
Human Dec. 2013 (GRCh38/hg38)(hg38)The exact name of this version of the human reference genome may vary between Galaxy servers. Start typing
hg38in the select box to reveal available choices on your server.- “Single or Paired-end reads”:
Paired Collection
- “Select a paired collection”: the
pairedoutput of Trimmomatic
Obtaining the identifiers of non-human read pairs
The mapping step above has produced a collection of BAM datasets that store, for each sample, which of its reads map to the human genome (and if so to which position on the genome though we are not interested in this information), and which ones could not be mapped to it. It is these unmapped reads that we are interested in, but how do we get at them?
The logic of the following steps is:
-
Filter the BAM dataset for each sample for only those reads that could not be mapped to the human reference genome, and for which also the read mate in the pair could not be mapped to this genome.
In other words, if only one read of a read pair can be mapped to the human genome we play it safe and discard the pair.
-
Emit the remaining reads in a format (we choose FASTA here) from which it is easy to extract the read identifiers
-
Extract the read identifiers
Hands On: Filter for read pairs not mapping to the human genome and extract their identifiers
- Samtools fastx ( Galaxy version 1.9+galaxy1) with the following parameter settings:
- param-collection “BAM or SAM file to convert”: the output of Map with BWA-MEM
- “Output format”:
FASTA“outputs”: select
READ1With this setting we are choosing to retrieve only the forward reads of each pair as a single result dataset. Since the forward and reverse reads of a pair share their read identifier, we do not need the reverse reads.
“Omit or append read numbers”:
Do not append /1 and /2 to read namesAdding this first or second read of the pair indicator to the read name would make the result dependent on which half of the reads we retrieve. We do not want this.
- “Require that these flags are set”:
read is unmappedmate is unmappedThis setting makes sure we retain only read pairs for which none of the two reads could be mapped to the human genome.
- Select lines that match an expression with the following parameters:
- param-collection “Select lines from”: the output of Samtools fastx
- “that”:
Matching- “the pattern”:
^>.+This will select only those lines from the FASTA inputs that start with a
>character, i.e. will retain the identifier lines, but not the sequence lines.- Replace Text in entire line ( Galaxy version 1.1.2) with the following parameters:
- param-collection “File to process”: the output of Select
- param-repeat “Replacement”:
- “Find Pattern”:
^>(.+)- “Replace with”:
\1This find and replacement pattern will work on lines starting with a
>character and replace each such line with the content following the>character (the(.+)part that gets referenced as\1in the replacement pattern).
Use the non-human read identifiers to extract the reads of interest from the original inputs
We are almost there! We already know the identifiers of all reads we would like to retain, we only need a way to filter the original input datasets for these identifiers.
Comment: Why not use the unmapped reads directly?When you followed the previous steps you may have noticed that Samtools fastx offers fastq as an output format. Since we used this tool to report the unmapped reads, you may be wondering why we did not simply ask the tool to write the reads in the format we want and be done with it.
The answer is: we could have done that (and some people follow this approach), but remember that we trimmed the reads before mapping them, which means the extracted unmapped reads would not be identical to the input reads.
It is good practice to submit sequencing data to public databases in as raw a state as possible to leave it up to the people downloading that data to apply processing steps as they see fit.
The seqtk_subseq tool offfers the identifier-based filtering functionality we are looking for. As a slight complication, however, the tool is not prepared to handle paired read collections like the one we organized our input data into. For this reason, we need to first unzip our collection into two simple list collections - one with the forward reads, the other one with the reverse reads of all samples. Then, after processing both with seqtk_subseq, we zip the filtered collections back into one.
Hands On: Identifier-based extraction of non-human reads from the input data
- Unzip Collection with the following setting:
- “Input Paired Dataset”: the collection of original sequenced reads input datasets
- seqtk_subseq ( Galaxy version 1.3.1) with the following parameters:
- param-collection “Input FASTA/Q file”: the first (forward reads) output of Unzip Collection
- “Select source of sequence choices”:
FASTA/Q ID list
- param-collection the collection of read identifiers; output of Replace Text
- seqtk_subseq ( Galaxy version 1.3.1) with the following parameters:
- param-collection “Input FASTA/Q file”: the second (reverse reads) output of Unzip Collection
- “Select source of sequence choices”:
FASTA/Q ID list
- param-collection the collection of read identifiers; output of Replace Text
- Zip Collection with the following settings:
- param-collection “Input Dataset (Forward)”: the collection of cleaned forward reads; output of the first run of seqtk_subseq
- param-collection “Input Dataset (Reverse)”: the collection of cleaned reverse reads; output of the second run of seqtk_subseq
Conclusion
Data cleaning is a standard procedure for clinical sequencing datasets and is a rather straightforward process in Galaxy. You just learnt how to remove human reads from any number of paired-end sequenced samples in parallel using collections and just a handful of tools. Cleaning of single-end data would use the same tools (with adjusted settings), but would work on data arranged in a simple list collection instead of in a paired collection.
You've Finished the Tutorial
Key points
Before submitting raw viral sequencing data to public databases you will want to remove human sequence traces
Human reads can be identified by mapping the data to the human reference genome
After mapping, you can extract the read identifiers of the non-human reads and use these to extract just the desired original sequenced reads from the input data
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Wolfgang Maier, Removal of human reads from SARS-CoV-2 sequencing data (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/human-reads-removal/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{sequence-analysis-human-reads-removal, author = "Wolfgang Maier", title = "Removal of human reads from SARS-CoV-2 sequencing data (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/human-reads-removal/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/sequence-analysis/tutorials/human-reads-removal/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bwa owner: devteam revisions: e188dc7a68e6 tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_fastx owner: iuc revisions: a8d69aee190e tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: seqtk owner: iuc revisions: 3da72230c066 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: trimmomatic owner: pjbriggs revisions: 898b67846b47 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: trimmomatic owner: pjbriggs revisions: d94aff5ee623 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/