Frequently Asked Questions
Format
FASTQ format
Although it looks complicated (and maybe it is), the FASTQ format is easy to understand with a little decoding. Each read, representing a fragment of DNA, is encoded by 4 lines:
Line Description 1 Always begins with @followed by the information about the read2 The actual nucleic sequence 3 Always begins with a +and contains sometimes the same info in line 14 Has a string of characters which represent the quality scores associated with each base of the nucleic sequence; must have the same number of characters as line 2 So for example, the first sequence in our file is:
@03dd2268-71ef-4635-8bce-a42a0439ba9a runid=8711537cc800b6622b9d76d9483ecb373c6544e5 read=252 ch=179 start_time=2019-12-08T11:54:28Z flow_cell_id=FAL10820 protocol_group_id=la_trappe sample_id=08_12_2019
AGTAAGTAGCGAACCGGTTTCGTTTGGGTGTTTAACCGTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTCGTGCGGAAGGCGCTTCACCCAGGGCCTCTCATGCTTTGTCTTCCTGTTTATTCAGGATCGCCCAAAGCGAGAATCATACCACTAGACCACACGCCCGAATTATTGTTGCGTTAATAAGAAAAGCAAATATTTAAGATAGGAAGTGATTAAAGGGAATCTTCTACCAACAATATCCATTCAAATTCAGGCA
+
$'())#$$%#$%%'-$&$%'%#$%('+;<>>>18.?ACLJM7E:CFIMK<=@0/.4<9<&$007:,3<IIN<3%+&$(+#$%'$#$.2@401/5=49IEE=CH.20355>-@AC@:B?7;=C4419)*$$46211075.$%..#,529,''=CFF@:<?9B522.(&%%(9:3E99<BIL?:>RB--**5,3(/.-8B>F@@=?,9'36;:87+/19BAD@=8*''&''7752'$%&,5)AM<99$%;EE;BD:=9<@=9+%$It means that the fragment named
@03dd2268-71ef-4635-8bce-a42a0439ba9a(ID given in line1) corresponds to:
- the DNA sequence
AGTAAGTAGCGAACCGGTTTCGTTTGGGTGTTTAACCGTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTCGTGCGGAAGGCGCTTCACCCAGGGCCTCTCATGCTTTGTCTTCCTGTTTATTCAGGATCGCCCAAAGCGAGAATCATACCACTAGACCACACGCCCGAATTATTGTTGCGTTAATAAGAAAAGCAAATATTTAAGATAGGAAGTGATTAAAGGGAATCTTCTACCAACAATATCCATTCAAATTCAGGCA(line2)- this sequence has been sequenced with a quality
$'())#$$%#$%%'-$&$%'%#$%('+;<>>>18.?ACLJM7E:CFIMK<=@0/.4<9<&$007:,3<IIN<3%+&$(+#$%'$#$.2@401/5=49IEE=CH.20355>-@AC@:B?7;=C4419)*$$46211075.$%..#,529,''=CFF@:<?9B522.(&%%(9:3E99<BIL?:>RB--**5,3(/.-8B>F@@=?,9'36;:87+/19BAD@=8*''&''7752'$%&,5)AM<99$%;EE;BD:=9<@=9+%$(line 4).But what does this quality score mean?
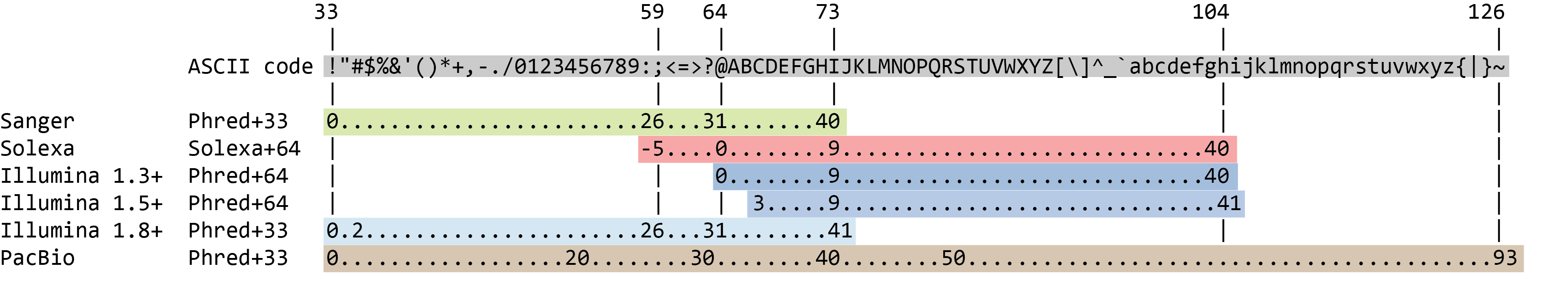
The quality score for each sequence is a string of characters, one for each base of the nucleotide sequence, used to characterize the probability of misidentification of each base. The score is encoded using the ASCII character table (with some historical differences):
So there is an ASCII character associated with each nucleotide, representing its Phred quality score, the probability of an incorrect base call:
Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% 60 1 in 1,000,000 99.9999%
Punteggi di qualità
Ma cosa significa questo punteggio di qualità?
Il punteggio di qualità per ogni sequenza è una stringa di caratteri, uno per ogni base della sequenza nucleotidica, utilizzata per caratterizzare la probabilità di errata identificazione di ogni base. Il punteggio è codificato utilizzando la tabella dei caratteri ASCII (con alcune differenze storiche):
Per risparmiare spazio, il sequenziatore registra un carattere ASCII per rappresentare i punteggi da 0 a 42. Ad esempio, 10 corrisponde a “+”. Ad esempio, 10 corrisponde a “+” e 40 a “I”. FastQC sa come tradurlo. Questo viene spesso chiamato punteggio “Phred”.
Quindi a ogni nucleotide è associato un carattere ASCII che rappresenta il suo punteggio di qualità Phred, la probabilità di una chiamata di base errata:
Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% 60 1 in 1,000,000 99.9999% Cosa rappresenta 0-42? Questi numeri, se inseriti in una formula, ci dicono la probabilità di errore per quella base. Questa è la formula, dove Q è il nostro punteggio di qualità (0-42) e P è la probabilità di errore:
Q = -10 log10(P)Utilizzando questa formula, possiamo calcolare che un punteggio di qualità di 40 significa solo 0,00010 probabilità di errore!
Puntuación de calidad
¿Pero qué significa esta puntuación de calidad?
La puntuación de calidad de cada secuencia es una cadena de caracteres, uno por cada base de la secuencia de nucleótidos, que se utiliza para caracterizar la probabilidad de identificación errónea de cada base. La puntuación se codifica utilizando la tabla de caracteres ASCII (con algunas diferencias históricas):
Para ahorrar espacio, el secuenciador registra un carácter ASCII para representar las puntuaciones 0-42. Por ejemplo, 10 corresponde a “+” y 40 a “I”. FastQC sabe cómo traducir esto. A menudo se denomina puntuación “Phred”.
Así que hay un carácter ASCII asociado a cada nucleótido, que representa su puntuación de calidad Phred, la probabilidad de una llamada de base incorrecta:
Puntuación de calidad Phred Probabilidad de una llamada de base incorrecta Precisión de llamada de base 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% 60 1 in 1,000,000 99.9999% ¿Qué representa 0-42? Estos números, cuando se introducen en una fórmula, nos indican la probabilidad de error para esa base. Esta es la fórmula, donde Q es nuestra puntuación de calidad (0-42) y P es la probabilidad de error:
Q = -10 log10(P)Utilizando esta fórmula, podemos calcular que una puntuación de calidad de 40 significa sólo 0,00010 de probabilidad de error
Quality Scores
But what does this quality score mean?
The quality score for each sequence is a string of characters, one for each base of the nucleotide sequence, used to characterize the probability of misidentification of each base. The score is encoded using the ASCII character table (with some historical differences):
To save space, the sequencer records an ASCII character to represent scores 0-42. For example 10 corresponds to “+” and 40 corresponds to “I”. FastQC knows how to translate this. This is often called “Phred” scoring.
So there is an ASCII character associated with each nucleotide, representing its Phred quality score, the probability of an incorrect base call:
Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% 60 1 in 1,000,000 99.9999% What does 0-42 represent? These numbers, when plugged into a formula, tell us the probability of an error for that base. This is the formula, where Q is our quality score (0-42) and P is the probability of an error:
Q = -10 log10(P)Using this formula, we can calculate that a quality score of 40 means only 0.00010 probability of an error!
Qualitätswerte
Aber was bedeutet dieser Qualitätswert?
Der Qualitätsscore für jede Sequenz ist eine Zeichenkette, eine für jede Base der Nukleotidsequenz, um die Wahrscheinlichkeit einer falschen Identifizierung jeder Base zu charakterisieren. Der Score wird unter Verwendung der ASCII-Zeichentabelle (mit einigen historischen Unterschieden) kodiert:
Um Platz zu sparen, zeichnet der Sequenzer ein ASCII-Zeichen auf, um die Punktzahlen 0-42 darzustellen. Zum Beispiel entspricht 10 einem “+” und 40 einem “I”. FastQC weiß, wie dies zu übersetzen ist. Dies wird oft als “Phred”-Bewertung bezeichnet.
Jedem Nukleotid ist also ein ASCII-Zeichen zugeordnet, das seinen Phred-Qualitätsscore, die Wahrscheinlichkeit eines falschen Basenrufs, darstellt:
Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% 60 1 in 1,000,000 99.9999% Was bedeutet 0-42? Wenn man diese Zahlen in eine Formel einsetzt, erhält man die Fehlerwahrscheinlichkeit für diese Basis. Die Formel lautet wie folgt: Q ist unser Qualitätswert (0-42) und P ist die Fehlerwahrscheinlichkeit:
Q = -10 log10(P)Mit dieser Formel können wir berechnen, dass eine Qualitätsbewertung von 40 nur 0,00010 Wahrscheinlichkeit eines Fehlers bedeutet!
Visualisation
Using IGV with Galaxy
You can send data from your Galaxy history to IGV for viewing as follows:
- Install IGV on your computer (IGV download page)
- Start IGV
- In recent versions of IGV, you will have to enable the port:
- In IGV, go to
View > Preferences > Advanced- Check the box
Enable Port- In Galaxy, expand the dataset you would like to view in IGV
- Make sure you have set a reference genome/database correctly (dbkey) (instructions)
- Under
display in IGV, click onlocal