Raw reads contain valuable information, such as coverage depth and quality scores, that is lost in a consensus sequence.
Submission of raw reads to public repositories allows reuse of data and reproducibility of analysis and enables discovery of minor allelic variants and intrahost variation, for example during the recent COVID-19 pandemic (Maier et al. 2021).
The European Nucleotide Archive is an Open and FAIR repository of nucleotide data. As part of the International Nucleotide Sequence Database Collaboration (INSDC), ENA also indexes data from the NCBI and DDBJ Arita et al. 2020. Data submitted to ENA must be accompanied by sufficient metadata. You can learn more from this introductory slide deck or directly from ENA.
In this tutorial we will show you how to use Galaxy’s ‘ENA Upload tool’ to submit raw sequencing reads, consensus sequences and their associated metadata to ENA (Roncoroni et al. 2021). You will learn to add your ENA Webin credentials to Galaxy, input metadata interactively or via a metadata template and submit the reads to ENA (test) server using Galaxy’s ‘ENA upload tool’.
Specifically, we will use one ONT sequencing file to demonstrate interactive metadata input and two sets of PE Illumina reads to demonstrate how to use the ENA metadata template. Finally, we will submit consensus sequences to ENA using ‘Submit consensus sequence to ENA’ tool.
Data will be submitted to ENA’s test server and will not be public.
Comment: Nature of the input data
We will use data derived from sequencing data of bronchoalveolar lavage fluid (BALF) samples obtained from early COVID-19 patients in China as our input data.
Human traces have been removed in Galaxy.
Adding ENA Webin credentials to your Galaxy user information
In order to submit data to ENA, you need to have a valid Webin account. If you don’t have one already you can register for one on ENA’s Registration Page. Webin credentials need to be included in your Galaxy user information before you can use the ENA Upload tool.
Hands On: Add Webin credentials to your Galaxy user information
If you have not already done so, log in to usegalaxy.eu
Navigate to “User” > “Preferences” on the top menu
Click on Manage Information
Scroll down to “Your ENA Webin account details” and fill in your ENA Webin ID and Password
Option 1: submitting to ENA using interactive metadata generator
In this first example, you will submit one ONT sequence file using the interactive metadata forms from the ENA Upload tool. This method is only convenient for small submissions. For bulk submissions, we recommend you use the metadata template described below in Option 2.
Hands On: Data upload
Upload the ONT data from Zenodo via URLs
Copy the link location
Click galaxy-uploadUpload at the top of the activity panel
Once the data is uploaded, we fill the metadata using the ENA Upload tool. Interactive metadata forms are nested to fit ENA’s metadata model. Briefly, you add Samples to a Study, Experiments to Samples and Runs to Experiments. The interactive metadata form does only two ENA Sample Checklists, the basic minimal sample metadata and the ENA virus pathogen reporting standard checklist. Switch between the basic template and the virus pathogen one under the “Select your sample type” option. If you wish to include additional metadata from a sample checklist, please use Option 2 below.
We recommend always submitting to the test server before submitting to the public one.
After you confirm that all the data and metadata looks ok, you can go ahead and submit to the public ENA server.
Hands On: add metadata interactively and submit a single sequence to ENA
ENA Upload tool ( Galaxy version 0.7.1):
“Action to execute”: Add new (meta)data
“Select the metadata input method”: Interactively (only recommended for small studies)
“Add .fastq (.gz, .bz2) extension to the Galaxy dataset names to match the ones described in the input tables?”: No
“Select your sample type”: Viral
Under “Submission options”:
“Affiliation centre”: your institution
“Submit to ENA test server”: yes
“Create test outputs without submitting (meta)data to ENA”: no
Fill all metadata boxes and make sure that:
”“:
“Please select the type of study”: Whole Genome Sequencing
“Enter the species of the sample”: Severe acute respiratory syndrome coronavirus 2
“Enter the taxonomic ID corresponding to the sample species”: 2697049
“Host common name”: human
“Host subject id”: avoid using ID that can be use to trace samples back to patients
“Host scientific name”: Homo sapiens
“Library strategy”: RNA-Seq
“Select library source”: METAGENOMIC
“Library selection”: RANDOM
“Library layout”: SINGLE
“Select the sequencing platform used”: Oxford Nanopore
“Instrument model”: minION
“Runs executed within this experiment”
param-files“File(s) associated with this run”: SRR10902284_ONT.fq.gz
Warning: Do not include personal identifiable data
In some cases, some information is requested by ENA that may classify as personal or could be used to identify persons (e.g. ‘host ID’ for checklist ERC000033). Make sure that you do not publish any personal metadata that infringes privacy protection regulations in your jurisdiction.
Warning: Submit to the test server first
Make sure “Submit to ENA test server”: yes. Otherwise your data will be submitted to the public server.
Four metadata tables (Study, Sample, Experiment and Run), and a metadata ticket with submission information are generated. You can confirm a successful submission at ENA test server (or the public server, if you chose it).
Upon successful submission, a metadata ticket is generated. This contains information of the submission, including parseable metadata. Importantly, it contains Study, Sample, Run and Experiment accession numbers. The former two you will use later to link the consensus sequence to the raw data.
Option 2: submitting to ENA using a metadata template
For larger submissions, interactive metadata input can be tedious and not practical. In the second example, you will submit two sets of Illumina PE sequence files and input metadata using a template spreadsheet. This template contains all fields for ENA sample checklist ‘ERC000033 - ENA virus pathogen reporting standard checklist’. Tabular (.tsv and .xlsx) metadata templates for all sample checklist can be found in this repository.
For this tutorial, we provide you with a pre-filled template and encourage you to explore it.
Click on galaxy-selectorSelect Items at the top of the history panel
Check all the datasets in your history you would like to include
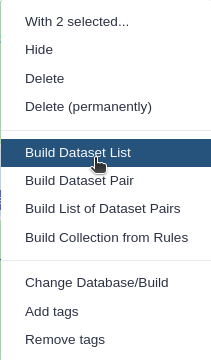
Click n of N selected and choose Advanced Build List
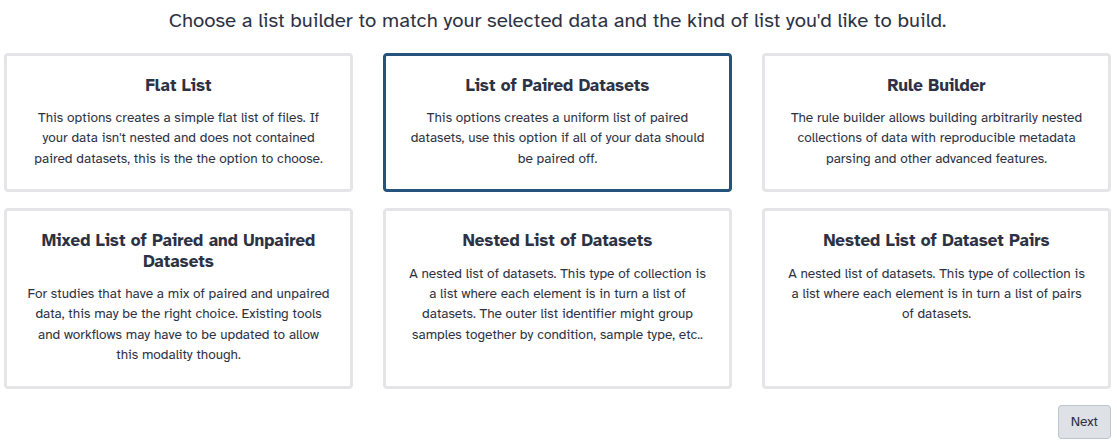
You are in the collection building wizard. Choose List of Paired Datasets and click ‘Next’ button at the right bottom corner.
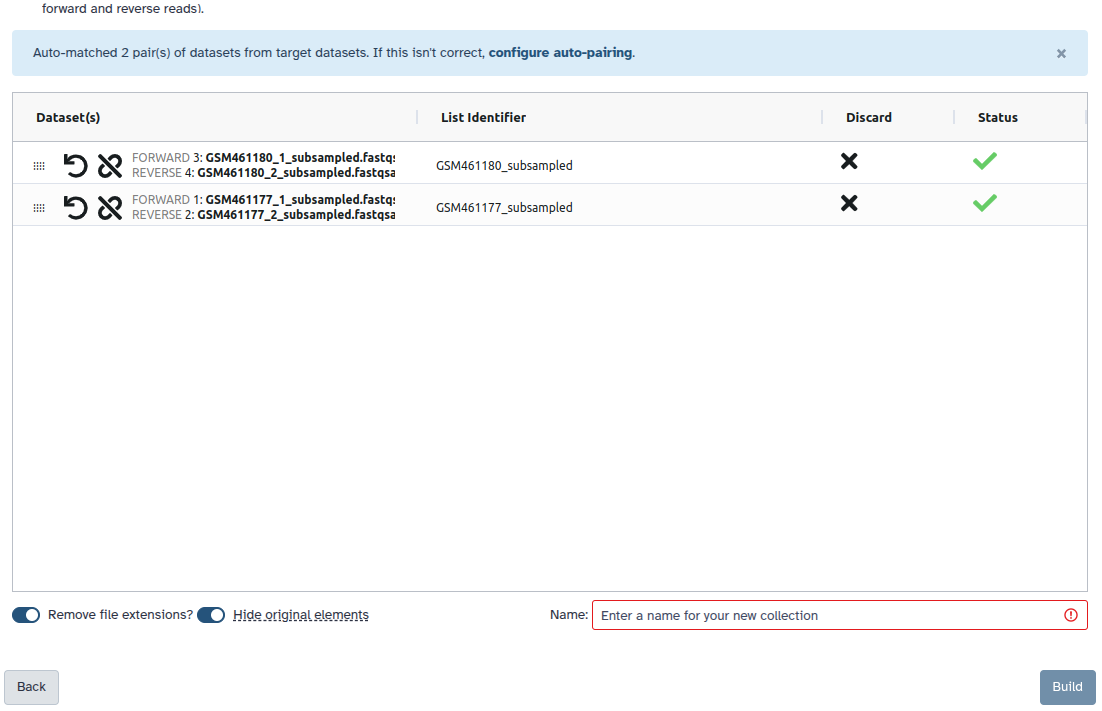
Check and configure auto-pairing. Commonly matepairs have suffix _1 and _2 or _R1 and _R2. Click on ‘Next’ at the bottom.
Edit the List Identifier as required.
Enter a name for your collection
Click Build to build your collection
Click on the checkmark icon at the top of your history again
For the example datasets this means:
You need to tell Galaxy about the suffix for your forward and reverse reads, respectively:
set the text of unpaired forward to: _1.fastq.gz
set the text of unpaired reverse to: _2.fastq.gz
click: Auto-pair
All datasets should now be moved to the paired section of the dialog, and the middle column there should show that only the sample accession numbers, i.e.SRR10903401 and SRR10903402, will be used as the pair names.
Warning: Paired collection names
It is very important that the paired collection names contain no suffix (e.g. _1, _R1, etc.) or file extensions (.fastq, .fastq.gz). The submission tool will add these at runtime and leaving them in the paired collection names will cause a mismatch with the filenames in the metadata table.
Make sure Hide original elements is checked to obtain a cleaned-up history after building the collection.
Give your collection a name
Click Create Collection
Inspect the GTN_tutorial_mock_metadata.xlsx (filled-in template) file by clicking on the galaxy-eye (eye) icon
https://github.com/enasequence/webin-cli
Question
How many metadata sheets are there?
How is the ‘Sample’ section in the template different from that in the interactive metadata input?
There are four metadata sheets, one per metadata object (Study, Sample, Experiment, Run)
The Sample section is more extensive in the template spreadsheet, because it contains all the fields from ENA ERC000033 sample checklist, as well as all ‘Recommended’ and ‘Optional’ fields in the other sections.
As before, the submission is done to the test server before submitting to the public one.
Hands On: use a metadata template and submit multiple sequences to ENA
ENA Upload tool ( Galaxy version 0.7.1):
“Action to execute”: Add new (meta)data
“Select the metadata input method”: Excel file
“Select the ENA sample checklist”: ENA virus pathogen reporting standard checklist (ERC000033)
“Select Excel (.xlsx) file based on template”: metadata_template_ERC000033_mock_complete.xlsx
“Select input data”: Paired dataset collection
“List of paired-end sequencing data files”: select the collection you made above during data upload
Under “Submission options”:
“Affiliation centre”: your institution
“Submit to ENA test server?”: yes
“Create test outputs without submitting (meta)data to ENA”: no
Warning: Do not include personal identifiable data
In some cases, some information is requested by ENA that may classify as personal or could be used to identify persons (e.g. ‘host ID’ for checklist ERC000033). Make sure that you do not publish any personal metadata that infringes privacy protection regulations in your jurisdiction.
Warning: Submit to the test server first
Make sure “Submit to ENA test server?”: yes. Otherwise your data will be submitted to the public server.
Four metadata tables (Study, Sample, Experiment and Run), and a metadata ticket with submission information are generated. You can confirm a successful submission at ENA test server (or the public server, if you chose it).
Upon succesful submission, a metadata ticket is generated. This contains information of the submission, including parseable metadata. Importantly, it contains Study, Sample, Run and Experiment accession numbers. The former two you will use later to link the consensus sequence to the raw data.
In this step we will submit one consensus sequence to ENA.
We will link it to the reads submitted in the first step using the accession numbers given to those submissions. Galaxy’s ‘ENA Upload tool’ captures and stores metadata on a metadata ticket.
Hands On: submit consensus sequences to ENA
Upload the consensus sequence to Galaxy from Zenodo via URLs:
galaxy-eye Open the ‘ENA submsission receipt’ and find the Study and Sample accession numbers from the raw data submission.
Submit consensus sequence to ENA ( Galaxy version 4.3.0):
“Submit to test ENA server?”: yes
“Validate files and metadata but do not submit”: no
Fill the assembly metadata. For our assembly:
“Assembly type”: Clone
“Assembly program”: BWA-MEM
“Molecule type”: genomic RNA
“Coverage”: 1000
“Select the method to load study and sample metadata”: Fill in required metadata
“Assembly name”: give a name to your assembly.
“Study accession”: ERP139884 (you can find the Study accession number from your raw data submission metadata ticket)
“Sample accession”: ERS12519941 (you can find the Sample accession number from your raw data submission metadata ticket)
“Sequencing platform”: Illumina
Select the consensus sequence assembly file from your history: SRR10903401.fasta
The output are a list of manifest (in our example one manifest) and a ‘submission log’. The manifest file is the metadata required by ENA’s Webin CLI tool. The ‘submission log’ can be useful to troubleshoot failed submissions.
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
Use Galaxy’s ‘ENA Upload tool’ to submit raw reads to ENA
Use Galaxy’s ‘Submit consensus sequence to ENA’ tool
You need to include your ENA Webin credentials in Galaxy
For small submission use ‘ENA Upload tool’ interactive metadata forms feature
For bulk submissions use a spreadsheet metadata template
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channels
Arita, M., I. Karsch-Mizrachi, and G. Cochrane, 2020 The international nucleotide sequence database collaboration. Nucleic Acids Research 49: D121–D124. 10.1093/nar/gkaa967
Maier, W., S. Bray, M. van den Beek, D. Bouvier, N. Coraor et al., 2021 Freely accessible ready to use global infrastructure for SARS-CoV-2 monitoring. 10.1101/2021.03.25.437046
Roncoroni, M., B. Droesbeke, I. Eguinoa, K. D. Ruyck, F. D’Anna et al., 2021 A SARS-CoV-2 sequence submission tool for the European Nucleotide Archive (Z. Lu, Ed.). Bioinformatics. 10.1093/bioinformatics/btab421
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{galaxy-interface-upload-data-to-ena,
author = "Miguel Roncoroni and Bert Droesbeke and Boris Depoortere",
title = "Submitting sequence data to ENA (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/upload-data-to-ena/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
You can use Ephemeris's shed-tools install command to install the tools used in this tutorial.
Questions:
Open image in new tab