Sequence data submission to ENA
| Author(s) |
|
| Editor(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
I am new to sequence submission to a repository.
How do I submit sequences and metadata to ENA using the Webin portal and cURL?
Requirements:
To populate ENA metadata objects through the Webin portal
To submit raw reads to ENA using FTP
- Web browser
- A linux-based machine or linux emulator
- Read data in fastq format
Time estimation: 1 hourSupporting Materials:Published: Nov 1, 2023Last modification: Jun 25, 2024License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00369rating Rating: 4.0 (0 recent ratings, 3 all time)version Revision: 5
DNA sequencing has become one of the key technologies in molecular biology, with applications in diagnostics, evolutionary biology, drug discovery, forensics and much more. Drop in sequencing costs and breakthroughs in sequencing technologies has seen increasing utilization of sequencing as a research tool, featuring in thousands of life-science publications every year.
Prior to publication many journals and funders require authors to submit their raw sequence data to one of the three INSDC member databases – ENA, NCBI or DDBJ – between which data is synchronised on a daily basis. INSDC is the core infrastructure for sharing nucleotide sequence data and metadata in the public domain. Data in INSDC member databases is available permanently, for free and with unrestricted access. For each submitted sequence a unique accession number is issued which can be reported in the publication.
The three databases have different methods for making submissions. If your database of choice is ENA and you need to submit data stored on a remote server, you are in the right place. This tutorial will cover how to find your way around the ENA Webin portal for uploading raw sequencing read data as well as accompanying metadata, and use cURL to copy read files over to ENA’s FTP server.
If you would like to use Galaxy tools for submission to ENA you may find Submitting sequence data to ENA tutorial helpful.
cURL is a command-line tool and library for transferring data over the internet. It allows you to send and receive data from various protocols like HTTP, FTP, and more. In simple terms, it’s a tool that helps your computer talk to other computers on the internet and fetch or send information, like downloading files from a website or making API requests.
AgendaIn this tutorial we will deal with:
ENA Submission Routes
There are three routes via which one can submit data to ENA.
- The Webin Interactive portal allows you to submit data by filling out forms on your browser. This is what we will cover in this tutorial,
- Webin-command-line interface (CLI), which allows submissions through a terminal, and
- Programmatic submission, which requires the submissions to be prepared as XML documents and then uploaded using cURL or Webin portal.
Table 1. ENA submission routes
Interactive Webin-CLI Programmatic Study Y N Y Sample Y N Y Read data Y Y Y Genome Assembly N Y N Transcriptome Assembly N Y N Template Sequence N Y N Other Analyses N N Y
As you can see from the Table 1, you can submit some data types only through a certain route. However, read data can be submitted via all three routes.
To begin with the submission process you will need to have a Webin submission account. You can register an account free of cost through the Webin portal (https://www.ebi.ac.uk/ena/submit/webin/login).
For data submission, ENA provides a detailed guide (https://ena-docs.readthedocs.io/en/latest/submit/general-guide.html), which I recommend you consult for questions specific to your user case. Remember that ENA frequently changes its submission protocols, so you should check the ENA guide for updates.
ENA Metadata Model
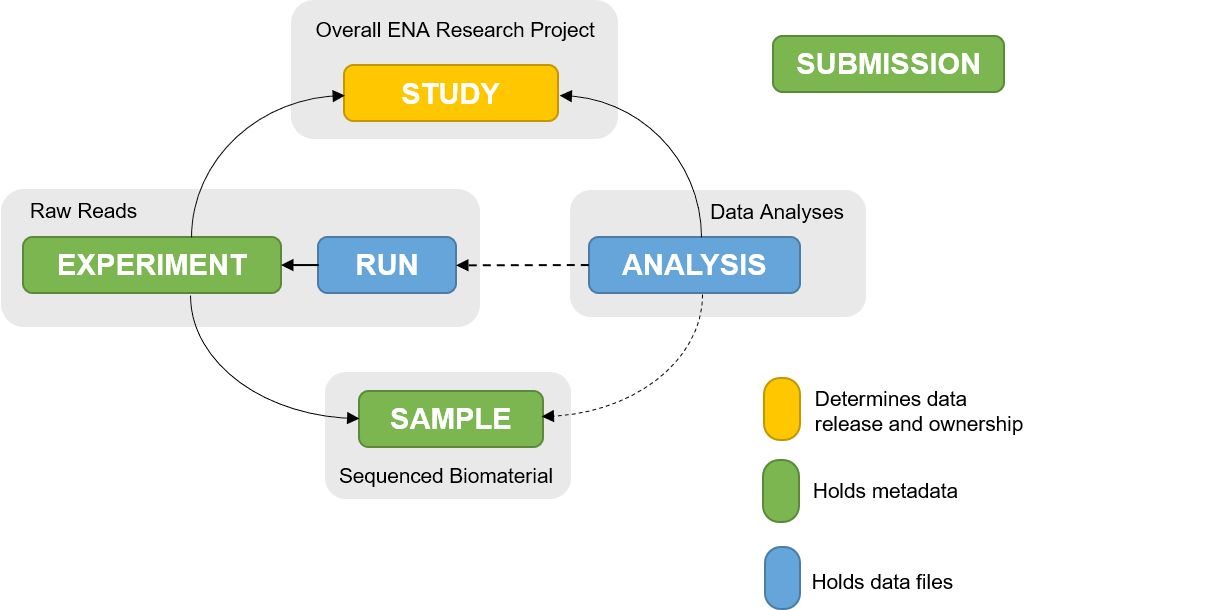 Open image in new tab
Open image in new tabThe ENA metadata model (Figure 1) represents how metadata for a submission is structured and linked. Different information pertaining to a submission is organised into objects within the metadata model. Therefore, understanding the metadata model will enable you to determine which objects you need to submit.
1. Study
All submissions require the study object. This object defines study accession, ownership, affiliation and release date. It also binds together all related objects into one cohesive project.
Study object submission generates a BioProject accession (PRJEB) which can be used for citing data submitted to ENA, and a study accession (ERP).
2. Sample
Sample object defines metadata about the sequenced biomaterial, e.g. bacteria, virus, etc. One sample should represent one sequencing library.
To ensure that each sample is registered with sufficient metadata so as to provide enough context for the data to be easily interpreted, a mandatory, recommended and optional set of attributes should be provided for a sample. Sample checklists have been developed in collaboration with different research communities to ensure that they are relevant and realistic for their context. They differ with the nature of the sample.
A registered sample will receive a BioSample accession (SAMEA) and ENA sample accession (ERS).
3. Raw reads
Metadata for raw reads is represented in ‘experiment’ and ‘run’ objects, where ‘experiment’ holds metadata on the sequencing method and ‘run’ holds metadata about the read files and their location on an FTP server.
Each read file submission will receive a run accession (ERR) and an Experiment accession (ERX).
02. Submitting data
1. Webin login
Log in to ENA Webin portal. If you don’t have a login you will have to register for an account. Go ahead and do that - it is free. Click on the ‘Register’ button and fill in the information requested.
2. Study metadata
Metadata preparation
To register a study you will need the following metadata:
- Study Name
- Short descriptive study title
- Detailed study abstract
- Release date (if before two years from the date of submission)
1-3 will be displayed on the Project entry page. Make it as detailed as possible so as to make your study searchable. To see an example see the submission for PRJEB55803.
Hands On: Register studyOnce you have the above information at hand register your study.
- From the Webin dashboard click on ‘Register Study’ to bring up the study submission form.
- Complete the form to your best knowledge - ensure attributes with ‘*’ are completed.
- Click ‘Submit’ to create a new study.
- If successful, you will receive a confirmational pop-up message with two accession numbers - PRJEB* (BioProject accession) and ERP*. Save them as you will need them for read submission step.
- Check the processing status of your submissions via the Studies Report.
You may want to set the submission date to the maximum allowed - 2 years from today. You can change it when you are ready to release the data. Study Fields are editable after submission should you need to update any information.
QuestionHow can you confirm that your study has been successfully registered?
Click on the Dashboard icon, select ‘Studies Report’. The accessions for your study should appear next to the Study title and other attributes.
3. Sample metadata
Next, you should register the samples from which your sequence data is derived.
Metadata preparation
Minimum metadata requirement for different checklists varies. We list only what is required for the ENA default checklist (ERC000011).
- Taxonomy ID of the organism sequenced
- Scientific name of the organism sequenced
- Unique name of the sample (auto-generated unless provided)
- Sample title
- Sample description
- Collection date
- Geographic location
Other checklists include:
- Pathogen checklist
- Environmental checklist
- Marine checklist
Find the complete list of checklists.
Hands On: Download sample checklist
- From the Dashboard, inside the ‘Samples’ box, click on ‘Register Samples’.
- Expand the ‘Download spreadsheet to register samples’ box and select ‘Other Checklists’.
- Click on ‘ENA default sample checklist’ (or the most appropriate checklist for your study).
- The mandatory attributes will be auto-selected. Expand the ‘Optional Fields’ box and select attributes you would like to include.
Your checklist will contain columns shown in Table 2.
Table 2. Sample metadata checklist
Checklist ERC000011 ENA default sample checklist tax_id scientific_name sample_alias sample_title sample_description collection date geographic location (country and/or sea) #units When registering a sample, it is recommended that you provide as much metadata as possible so as to make your study more searchable and useable.
QuestionWhat format should the collection date be in? Read descriptions for other attributes to get a clear understanding of the attributes and permitted values.
Date/time in any of these formats is acceptable: 2008-01-23T19:23:10+00:00; 2008-01-23T19:23:10; 2008-01-23; 2008-01; 2008
- Click on ‘Next’ to get to the ‘Download TSV Template’ button.
Hands On: Upload sample checklist
- Open the Checklist_*.tsv file in a text editor of your choice e.g. MS Excel.
- Fill one sample per row, starting from row 4. Make sure you fill in all the columns.
- In the ‘#units’ row specify the unit of measure, if required.
- Make sure you save the checklist.tsv file in tab-delimited format with .tsv or .tab extension and not .xlsx or .csv.
- In the Webin Portal, select ‘Register Samples’ and then ‘Upload filled spreadsheet to register samples’. Upload your checklist.tsv file via ‘Choose File’ button.
- Click on ‘Submit’ to submit the checklist.
- If the structure of your checklist is correct and the upload is successful a pop-up message will appear with Sample accession numbers - SAMEA* (BioSample) and ERS*.
- Check the processing status of your submissions via Samples Report.
Remember to enter sample aliases that correspond to what you use in related publications. This will enable readers to find sample-specific metadata and read files, even if you only state a Project accession number in your paper. Sample aliases can, furthermore, be optionally displayed as Unique name in the Sample report.
4. Read metadata
Metadata preparation
Having registered the sample, we can now add information about sequence reads generated from them. Reads can be submitted in several formats, such as fastq, BAM, CRAM, fast5, etc. we will look at submitting paired-end fastq files. Metadata required for this are:
- Sample alias or accession (from Sample submission step)
- Study alias or accession (from Study submission step)
- Sequencing instrument model (e.g. Illumina MiSeq, Illumina NextSeq 500, MinION, etc.)
- Library name (if any)
- Type of source material sequenced (E.g. genomic, transcriptomic, etc.)
- Library selection method used (e.g. PCR, cDNA, etc.)
- Library sequence strategy (e.g. RNA-Seq, WGS, etc.)
- Library layout i.e. single or paired-end
- Forward fastq file name (must be compressed, and name to include subdirectory)
- MD5 checksum of forward fastq file (if not using Webin file uploader)
- Reverse fastq file name (must be compressed, and name to include subdirectory)
- MD5 checksum of reverse fastq file (if not using Webin file uploader)
Fastq file preparation
First, we need to confirm that your read files are in the correct format. Refer to ENAs file format guidelines if you are not sure.
Hands On: Linux or OSXOn a Linux-based operating system
Compress the fastq files for the upload using gzip.
Open the terminal on your machine then type the commands below. First move to the directory where fastq files are located, then compress the fastq files using gzip command.
# In the command below replace '/path/to/fastq/directory' with the correct path cd /path/to/fastq/directory gzip *.fastqTo enable verification of the integrity of the uploaded fastq file, ENA requires md5 checksum for each file.
Type the command below to calculate and print md5 sums to tab-separated file (for easy cut-and-paste later).
for f in *.gz; do md5 $f | awk '{ gsub(/\(|\)/,""); print $2"\t" $4 }'; done > md5sums.tsvmd5sums.tsv will contain a tab-separated table of fastq.gz filenames and their md5sum.
Hands On: Read checklist submission
- From the ‘Dashboard’ select ‘Submit Reads’ from Raw Reads box.
- Similar to how you downloaded the sample checklist, download the Read submission template for ‘paired reads using two fastq files’.
- Select the relevant optional fields to be included in the spreadsheet.
- Tick the box next to ‘Show Descriptions’ to reveal text describing the attributes
- To find the sample alias and accession, look in ‘Sample Reports’
- To find the study accession, look in ‘Studies Reports’
- Click ‘Next’ and download the spreadsheet .tsv file.
- Fill in the spreadsheet, paying particular attention to the following: sample: same as you used in sample submission - alias or accession study: use study accession forward/reverse_file_name: ensure this is the same as what will be uploaded. It should be the compressed .gz file you created. forward/reverse_file_md5: copy this from md5sums.tsv you generated earlier.
- Uploaded the completed template spreadsheet and submitted it.
- If metadata validation is successful a pop-up message bearing a run accession (ERR) and experiment accession (ERX) will appear for each read submission.
- Confirm the processing status of your submission via Runs Report.
- Any errors generated in the process will be communicated to you on the registered email.
The run submission holds information about the raw read files generated in a run of sequencing as well as their location on an FTP server. The experiment submission holds metadata that describes the methods used to sequence the sample.
5. Read data upload
ENA offers several methods to achieve this. Please take a look at all the options should any suit your needs better than the one provided below.
If you are using a Linux-based system you can use the below commands without needing to install any software.
cd /path/to/fastq.gz
curl -T <<your_file>>.gz -u Webin-XXX:password ftp://webin2.ebi.ac.uk/
Check the processing status of your upload via the Run Processing Report.
WarningThe Webin upload area is a temporary transit location which is not backed up. Always ensure you retain a local copy of the data till the files have been successfully submitted and archived.
6. Post-submission editing
Metadata submitted for each object can be edited via the Reports (Studies, Samples, Runs) by clicking on the box-arrow icon in the Action column next to the table presenting the reports. The underlying xml of the submitted item can be inspected and edited.
However, for complex edits you might have to contact ENA helpdesk.
You've Finished the Tutorial
Key points
Use ENA Webin interactive portal to submit metadata
Preparation of metadata and sequence data for the submission
Use cURL to submit read fastq files
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsGlossary
- ENA
- European Nucleotide Archive
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Sonal Henson, Sequence data submission to ENA (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/fair/tutorials/fair-ena/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{fair-fair-ena, author = "Sonal Henson", title = "Sequence data submission to ENA (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/fair/tutorials/fair-ena/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!