Neoantigen prediction for HLA binding is a critical component of personalized cancer immunotherapy. Neoantigens, which are tumor-specific antigens resulting from mutations in cancer cells, can be recognized by the immune system, making them promising targets for tailored immunotherapies. Human leukocyte antigen (HLA) molecules play a key role in presenting these neoantigens on the surface of cells, where they can be detected by T-cells, triggering an immune response.
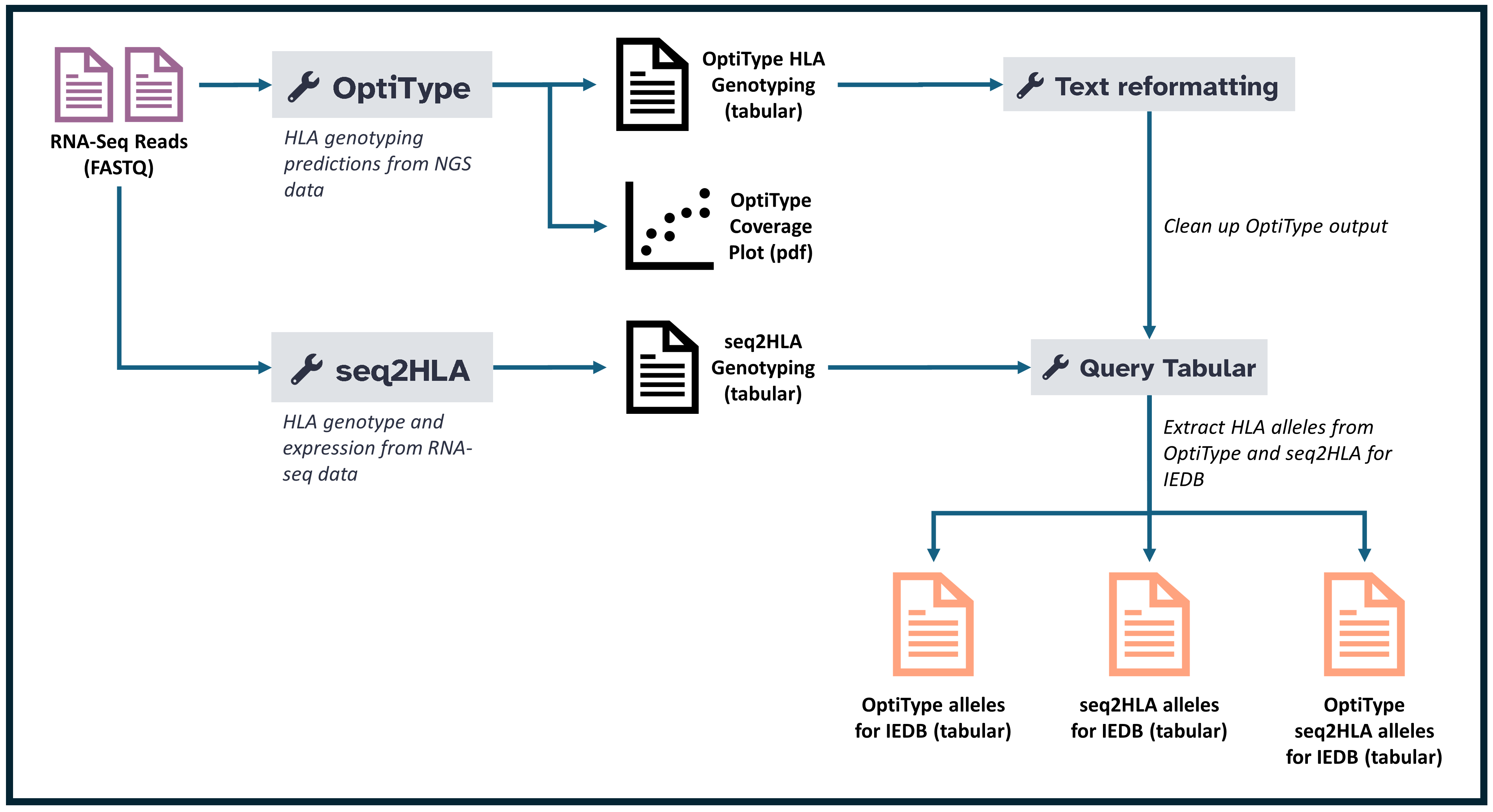
This tutorial focuses on predicting HLA binding affinities for potential neoantigens, an essential step in identifying effective targets for immunotherapy. The workflow includes the use of tools such as OptiType and seq2HLA to determine HLA genotypes, as well as data reformatting and querying methods to manage complex data outputs.
By completing this tutorial, learners will gain an understanding of:
The bioinformatics approaches required to predict HLA binding,
Practical skills for neoantigen identification, including managing and analyzing genomic data,
How to apply specific bioinformatics tools to analyze HLA binding predictions effectively.
This hands-on tutorial is designed to offer practical experience for learners aiming to integrate bioinformatics techniques into their immunotherapy research or clinical practice, emphasizing the theoretical and technical concepts essential for neoantigen prediction.
This tutorial provides a step-by-step guide for predicting HLA binding of neoantigens, a crucial part of personalized immunotherapy research. Using OptiType and seq2HLA, we will perform HLA typing and analyze which neoantigens are likely to bind to a specific individual’s HLA molecules, potentially driving an immune response. This process is essential for identifying candidate peptides that could serve as effective targets in immunotherapy.
Get Data The tutorial begins with the acquisition and upload of necessary sequencing files. These files typically contain raw paired-end reads or other sequencing outputs, which will be processed to identify HLA alleles. Proper organization and tagging of data are crucial for maintaining an efficient workflow.
HLA Typing with OptiType OptiType is used first in this tutorial for HLA typing. This tool processes the uploaded sequencing data to identify HLA class I alleles, which are essential for the subsequent neoantigen binding predictions. OptiType’s output will include the predicted HLA alleles that are specific to the individual’s immune profile.
HLA Typing with seq2HLA As a complementary approach, we will use seq2HLA for HLA typing to cross-validate results. By comparing predictions from both OptiType and seq2HLA, we can ensure greater accuracy in identifying relevant HLA alleles, allowing us to confidently proceed with neoantigen predictions.
Reformatting and Filtering HLA Alleles After obtaining results from both HLA typing tools, the next step is to reformat and filter the data. This involves removing redundant entries and preparing the data in a format that’s optimized for binding prediction. Cleaning and filtering ensure that only the most relevant HLA alleles are used in the neoantigen analysis.
Querying and Validating HLA Typing Results The final step is to use SQL-like queries to further refine and validate the HLA typing results. This allows us to filter and focus on the HLA alleles most relevant to our research question, ensuring the quality and relevance of the dataset for predicting neoantigen binding.
This structured workflow enables a streamlined approach for accurate HLA typing and binding prediction, setting the foundation for personalized immunotherapy and advancing the development of effective cancer treatments.
Get data
Hands On: Data Upload
Create a new history for this tutorial
Import the files from Zenodo or from
the shared data library (GTN - Material -> proteomics
-> Neoantigen 6: Predicting HLA Binding):
Click galaxy-uploadUpload Data at the top of the tool panel
Select galaxy-wf-editPaste/Fetch Data
Paste the link(s) into the text field
Press Start
Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
Go into Libraries (left panel)
Navigate to the correct folder as indicated by your instructor.
On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
Select the desired files
Click on Add to Historygalaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
“Select history”: the history you want to import the data to (or create a new one)
Click on Import
Rename the datasets
Check that the datatype
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, click galaxy-chart-select-dataDatatypes tab on the top
In the galaxy-chart-select-dataAssign Datatype, select datatypes from “New Type” dropdown
Tip: you can start typing the datatype into the field to filter the dropdown menu
Click the Save button
Add to each database a tag corresponding to …
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
Click on the dataset to expand it
Click on Add Tagsgalaxy-tags
Add tag text. Tags starting with # will be automatically propagated to the outputs of tools using this dataset (see below).
Press Enter
Check that the tag appears below the dataset name
Tags beginning with # are special!
They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
dataset 3 is used to calculate read coverage using BedTools Genome Coverageseparately for + and - strands. This generates two datasets (4 and 5 for plus and minus, respectively);
datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with #plus and #minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.
OptiType is a bioinformatics tool designed specifically for HLA class I typing using paired-end sequencing reads. In this workflow, OptiType identifies the HLA alleles that a person possesses, which are crucial for understanding which neoantigens may effectively bind to an individual’s immune receptors. This tool enhances the personalization of immunotherapy by providing highly accurate HLA allele predictions. In turn, this allows for more targeted analysis of potential neoantigen binding candidates.
Hands On: OptiType
OptiType ( Galaxy version 1.3.5+galaxy0) with the following parameters:
“Single or Paired-end reads”: Paired
param-file“Select first set of reads”: RNA-Seq_Reads_1.fastqsanger (Input dataset)
param-file“Select second set of reads”: RNA-Seq_Reads_2.fastqsanger (Input dataset)
“Enumerations”: 3
Question
Why do we use paired-end reads in OptiType?
What does the “Enumerations” parameter mean in OptiType, and why set it to 3?
Paired-end reads are used because they provide more comprehensive information about the DNA sequence. This improves the accuracy of HLA typing, as OptiType can analyze data from both directions of a DNA segment, reducing ambiguity and increasing the chance of correctly identifying HLA alleles.
The “Enumerations” parameter controls the number of best-matching HLA allele combinations that OptiType will report. Setting it to 3 provides multiple possible allele combinations for verification and ensures that we capture the most likely HLA types while still keeping the output manageable for downstream analysis.
HLA typing with seq2HLA
seq2HLA is a computational tool for identifying HLA types from RNA-Seq or DNA-Seq data. It is especially useful in immunogenomics research, as it allows for the prediction of an individual’s HLA class I and II alleles, which play a key role in immune response. In this workflow, seq2HLA provides essential data for identifying potential neoantigens by aligning sequencing reads to reference HLA alleles, allowing researchers to understand the specific immune profile of each sample.
Hands On: seq2HLA
seq2HLA ( Galaxy version 2.3+galaxy0) with the following parameters:
“Name prefix for this analysis”: STS26TGen
“Paired-end reads”: Paired
param-file“Select first set of reads”: RNA-Seq_Reads_1.fastqsanger (Input dataset)
param-file“Select second set of reads”: RNA-Seq_Reads_2.fastqsanger (Input dataset)
Question
Why is it important to specify a “Name prefix for this analysis”?
What is the significance of using paired-end reads in seq2HLA?
The “Name prefix for this analysis” parameter allows users to easily identify the output files related to a specific sample or experiment. By assigning a unique prefix, you can better organize and retrieve results, especially in workflows with multiple samples.
Paired-end reads enhance seq2HLA’s accuracy by providing complementary information from both ends of the DNA or RNA fragment. This improves the resolution and reliability of the HLA typing results, as more context is available to match against the reference HLA alleles.
Reformatting and Filtering HLA Alleles
This step involves reformatting and filtering the HLA alleles output from the OptiType tool to retain only the relevant and unique alleles. The process is essential for streamlining the data and ensuring that only the significant allele information is passed on for further analysis. Using a simple AWK program, we can filter out redundant data and focus on distinct HLA alleles, which are crucial for downstream neoantigen discovery and immunogenomics applications.
Hands On: Text reformatting
Text reformatting ( Galaxy version 9.3+galaxy1) with the following parameters:
param-file“File to process”: result (output of OptiTypetool)
“AWK Program”:
$1 ~ /[0-9]/{
for (i = 2; i <=7; i++) { allele[$i]++}
}
END {
for (i in allele) {
print i
}
}
Question
What is the purpose of using the AWK program in this step?
Why is it important to filter out redundant HLA alleles?
The AWK program here helps identify and filter unique HLA alleles from the output of OptiType. This step removes duplicates and ensures that only relevant alleles are retained for further analysis, improving data quality and accuracy.
Filtering out redundant alleles simplifies the dataset and enhances computational efficiency, making downstream analyses quicker and more focused on significant variants. This is essential for accurately interpreting immune responses in personalized medicine and neoantigen discovery.
Querying and Validating HLA Typing Results
In this step, we use the Query Tabular tool to validate and organize the HLA typing results from OptiType and seq2HLA outputs. By querying and filtering the data, we create a consolidated list of HLA alleles, formatted consistently for further analysis. This process ensures that we have a clean dataset of HLA alleles that will improve the accuracy of downstream analyses, such as neoantigen prediction or immune response studies.
Hands On: Query tabular
Query Tabular ( Galaxy version 3.3.2) with the following parameters:
In “Database Table”:
param-repeat“Insert Database Table”
param-file“Tabular Dataset for Table”: outfile (output of Text reformattingtool)
In “Filter Dataset Input”:
In “Filter Tabular Input Lines”:
param-repeat“Insert Filter Tabular Input Lines”
“Filter By”: by regex expression matching
“regex pattern”: ^(\w+[*]\d\d:\d\d\d?).*$
“action for regex match”: include line on pattern match
param-repeat“Insert Filter Tabular Input Lines”
“Filter By”: regex replace value in column
“enter column number to replace”: c1
“regex pattern”: ^(\w+[*]\d\d:\d\d\d?).*$
“replacement expression”: HLA-\1
In “Table Options”:
“Specify Name for Table”: optitype
param-repeat“Insert Database Table”
param-file“Tabular Dataset for Table”: c1_genotype4digits (output of seq2HLAtool)
Why is it necessary to use SQL queries on HLA typing results?
How do we know the query results are accurate for the next steps?
SQL queries allow us to filter, organize, and validate the HLA typing results efficiently. This approach ensures consistent formatting and removes unnecessary or redundant data, making the dataset more manageable for further analysis.
By carefully setting regex patterns and filtering options, we can ensure that only valid HLA allele formats are included. Reviewing the query results for consistency and completeness further ensures accuracy before moving to the next steps in the pipeline.
Conclusion
This tutorial covers the workflow for identifying and validating HLA alleles using OptiType and seq2HLA. Each step in the process, from reformatting and filtering to querying, is essential for generating accurate and consistent allele data. This validated data can then be used confidently in downstream immunological analyses, including neoantigen prediction and personalized medicine applications.
Rerunning on your own data
To rerun this entire analysis at once, you can use our workflow. Below we show how to do this:
Click on galaxy-workflows-activityWorkflows in the Galaxy activity bar (on the left side of the screen, or in the top menu bar of older Galaxy instances). You will see a list of all your workflows
Click on galaxy-uploadImport at the top-right of the screen
Paste the following URL into the box labelled “Archived Workflow URL”: https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/neoantigen-6-predicting-hla-binding/workflows/main_workflow.ga
Click the Import workflow button
Below is a short video demonstrating how to import a workflow from GitHub using this procedure:
Video: Importing a workflow from URL
Run Workflowworkflow using the following parameters:
Click on galaxy-workflows-activityWorkflows in the Galaxy activity bar (on the left side of the screen, or in the top menu bar of older Galaxy instances). At the top of the resulting page you will have the option to switch between the My workflows, Workflows shared with me and Public workflows tabs. Select the tab you want to see all workflows in that category.
Click on the workflow-runRun workflow button of the workflow you would like to use
Configure the workflow as needed
Click the Run Workflow button at the top-right of the screen
You may have to refresh your history to see the queued jobs
Disclaimer
Please note that all the software tools used in this workflow are subject to version updates and changes. As a result, the parameters, functionalities, and outcomes may differ with each new version. Additionally, if the protein sequences are downloaded at different times, the number of sequences may also vary due to updates in the reference databases or tool modifications. We recommend the users to verify the specific versions of software tools used to ensure the reproducibility and accuracy of results.
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
Neoantigen prediction is a key step in personalized medicine.
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{proteomics-neoantigen-6-predicting-hla-binding,
author = "Subina Mehta and Katherine Do and James Johnson",
title = "Neoantigen 6: Predicting HLA Binding (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/neoantigen-6-predicting-hla-binding/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
Do you want to extend your knowledge?
Follow one of our recommended follow-up trainings:
Questions: