Metabarcoding/eDNA through Obitools
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
how to analyze DNA metabarcoding / eDNA data produced on Illumina sequencers using the OBITools?
Requirements:
Deal with paired-end data to create consensus sequences
Clean, filter and anlayse data to obtain strong results
Time estimation: 1 hourSupporting Materials:Published: Feb 28, 2022Last modification: May 7, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00124rating Rating: 5.0 (0 recent ratings, 1 all time)version Revision: 13
Based on this OBITools official tutorial, you will learn here how to analyze DNA metabarcoding data produced on Illumina sequencers using:
- the OBITools on Galaxy
- some classical Galaxy tools
The data used in this tutorial correspond to the analysis of four wolf scats, using the protocol published in SHEHZAD et al. 2012 for assessing carnivore diet. After extracting DNA from the faeces, the DNA amplifications were carried out using the primers TTAGATACCCCACTATGC and TAGAACAGGCTCCTCTAG amplifying the 12S-V5 region (Riaz et al. 2011), together with a wolf blocking oligonucleotide.
It is always a good idea to have a look at the intermediate results or to evaluate the best parameter for each step. Some commands are designed for that purpose, for example you can use :
- obicount to count the number of sequence records in a file
- obihead and obitail to view the first or last sequence records of a file
- obistat to get some basic statistics (count, mean, standard deviation) on the attributes (key=value combinations) in the header of each sequence record (see The extended OBITools fasta format in the fasta format description)
- any Galaxy tools corresponding to classical unix command such as less, awk, sort, wc to check your files.
The OBITools programs imitate Unix standard programs because they usually act as filters. The main difference with classical Unix programs is that text files are not analyzed line per line but sequence record per sequence record (see below for a detailed description of a sequence record). Compared to packages for similar purposes like mothur (Schloss et al. 2009) or QIIME (Caporaso et al. 2010), the OBITools mainly rely on filtering and sorting algorithms. This allows users to set up versatile data analysis pipelines
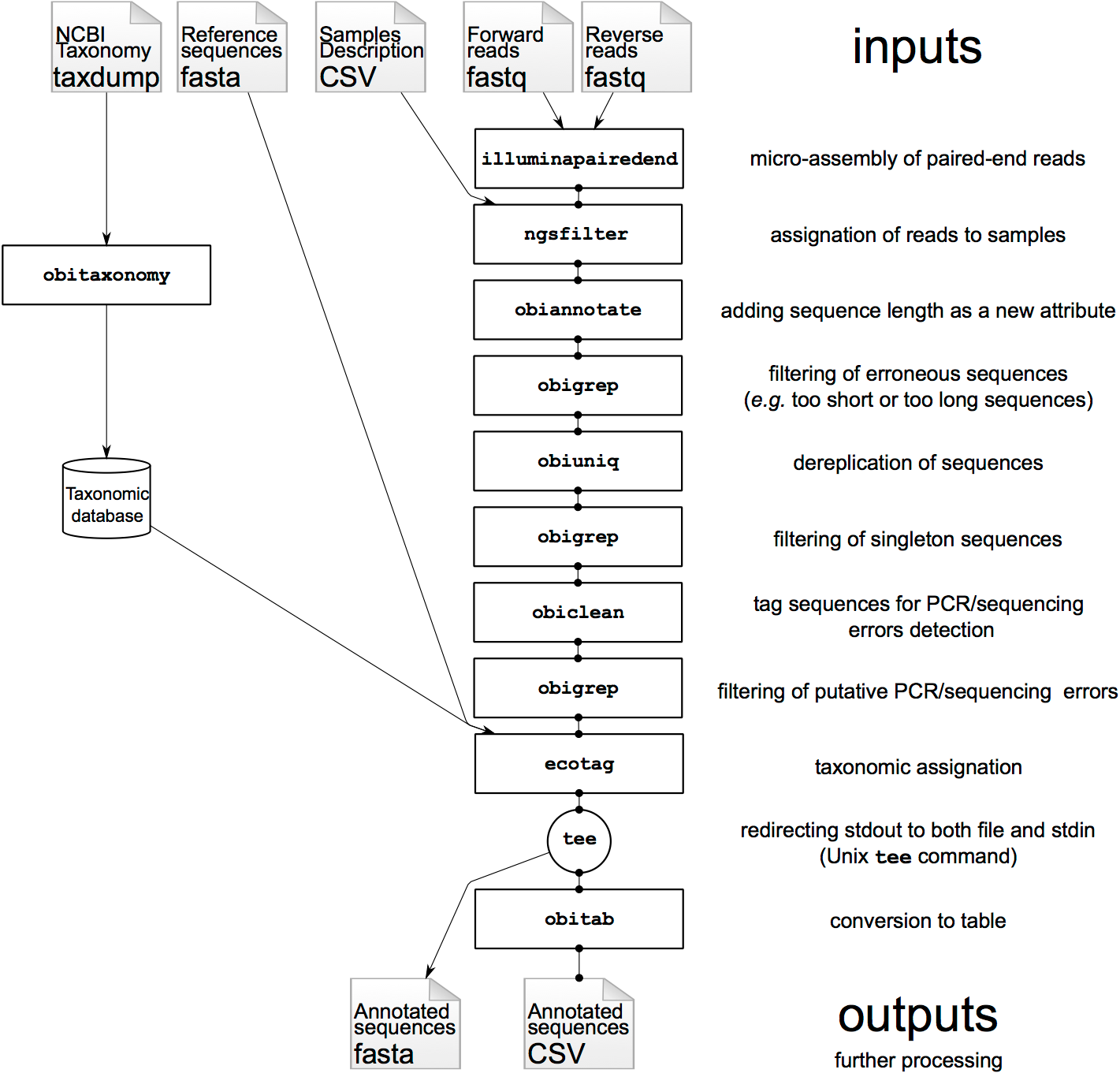
Most of the OBITools commands read sequence records from a file or from the stdin, make some computations on the sequence records and output annotated sequence records. As inputs, the OBITools are able to automatically recognize the most common sequence file formats (i.e. FASTA, FASTQ, EMBL, and GenBank). They are also able to read ecoPCR (Ficetola et al. 2010) result files and ecoPCR/ecoPrimers formatted sequence databases (Riaz et al. 2011) as ordinary sequence files. File format outputs are more limited. By default, sequences without and with quality information are written in FASTA and FASTQ formats, respectively. However, dedicated options allow enforcing the output format, and the OBITools are also able to write sequences in the ecoPCR/ecoPrimers database format, to produce reference databases for these programs. In FASTA or FASTQ format, the attributes are written in the header line just after the id, following a key=value; format.

AgendaIn this tutorial, we will cover:
- Manage input data
- Use OBITools
- Micro assembly of paired-end sequences with illuminapairedend
- Remove unaligned sequence records with obigrep
- Assign each sequence record to the corresponding sample/marker combination with NGSfilter
- Dereplicate reads into uniq sequences with obiuniq
- Limit number of informations with obiannotate
- Computes basic statistics for attribute values with obistat
- Filtering sequances by count and length with obigrep
- Clean the sequences for PCR/sequencing errors (sequence variants) with obiclean
- Taxonomic assignment of sequences with NCBI BLAST+ blastn
- Filter database and query sequences by ID to re associate informations with Filter sequences by ID
- From FASTA to tabular with Obitab
- create a final synthesis as a tabular file
- Conclusion
Manage input data
The data needed to run the tutorial are the following:
- FASTQ files resulting from a GA IIx (Illumina) paired-end (2 x 108 bp) sequencing assay of DNA extracted and amplified from four wolf faeces:
- wolf_F.fastq
- wolf_R.fastq
- the file describing the primers and tags used for all samples sequenced:
- wolf_diet_ngsfilter.txt
- the tags correspond to short and specific sequences added on the 5’ end of each primer to distinguish the different samples
- the file containing the reference database in a fasta format: -db_v05_r117.fasta This reference database has been extracted from the release 117 of EMBL using ecoPCR
Get data
Hands On: Data upload
- Create a new history for this tutorial
Import the zip archive containing input files from Zenodo
https://zenodo.org/record/5932108/files/wolf_tutorial.zip
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
- Rename the dataset, here a zip archive, if needed
Check that the datatype is
zip
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
datatypesfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button

Unzip the downloaded archive
Hands On: Unzip the downladed .zip archive and prepare unzipped files to be used by OBITools
- Unzip ( Galaxy version 0.2) with the following parameters:
- “Extract single file”:
All filesCommentTo work properly, this unzip Galaxy tool is waiting “simple” archive as input, this means without sub directory.
Add to each datafile a tag and/or modify names (optional)
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
- Click on the dataset to expand it
- Click on Add Tags galaxy-tags
- Add tag text. Tags starting with
#will be automatically propagated to the outputs of tools using this dataset (see below).- Press Enter
- Check that the tag appears below the dataset name
Tags beginning with
#are special!They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
- a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
- dataset 3 is used to calculate read coverage using BedTools Genome Coverage separately for
+and-strands. This generates two datasets (4 and 5 for plus and minus, respectively);- datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
- datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with
#plusand#minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.More information is in a dedicated #nametag tutorial.
Unhide all dataset from the resulting data collection so you can use these files independently.
- Modify datatype from txt to tabular for the
wolf_diet_ngsfilterdataset

Question
- Why do we need to unhide manually datasets from the data collection?
- Data collection is a functionality often used to deal with multiple datasets on the same format who can be analysed in batch mode. Here, the data collection is populated with heterogenous datafiles, coming from an archive. We thus need to treat separately each dataset of the collection, and to do so, we need to unhide corresponding datasets from the history, as datasets inside collections are just like “symbolic link” to “classical” history datasets hidden by default.
Use OBITools
OBITools (Boyer et al. 2015) is a set of programs specifically designed for analyzing NGS data in a DNA metabarcoding context, taking into account taxonomic information. It is distributed as an open source software available on the following website: http://metabarcoding.org/obitools.
The OBITools commands consider a sequence record as an entity composed of five distinct elements. Two of them are mandatory, the identifier (id) and the DNA or protein sequence itself. The id is a single word composed of characters, digits, and other symbols like dots or underscores excluding spaces. Formally, the ids should be unique within a dataset and should identify each sequence record unambiguously, but only a few OBITools actually rely on this property. The three other elements composing a sequence record are optional. They consist in a sequence definition, a quality vector, and a set of attributes. The last element is a set of attributes qualifying the sequence, each attribute being described by a key=value pair. The set of attributes is the central concept of the OBITools system. When an OBITools command is run on the sequence records included in a dataset, the result of the computation often consist in the addition of new attributes completing the annotation of each sequence record. This strategy of sequence annotation allows the OBITools to return their results as a new sequence record file that can be used as the input of another OBITools program, ultimately creating complex pipelines (source: OBITools Welcome).
Micro assembly of paired-end sequences with illuminapairedend
When using the result of a paired-end sequencing assay with supposedly overlapping forward and reverse reads, the first step is to recover the assembled sequence.
The forward and reverse reads of the same fragment are at the same line position in the two FASTQ files obtained after sequencing. Based on these two files, the assembly of the forward and reverse reads is done with the illuminapairedend utility that aligns the two reads and returns the reconstructed sequence.
Hands On: Recover consensus sequences from overlapping forward and reverse reads.
- illuminapairedend ( Galaxy version 1.2.13) with the following parameters:
- “Read from file”:
wolf_Ffor the 3p file- “Read from file”:
wolf_Rfor the 5p file- “minimum score for keeping aligment”:
40.0CommentSequence records corresponding to the same read pair must be in the same order in the two files !
If the alignment score is below the defined score, here 40, the forward and reverse reads are not aligned but concatenated, and the value of the mode attribute in the sequence header is set to joined instead of alignment
Remove unaligned sequence records with obigrep
In this step we are going to use the value of the mode attribute in the sequence header of the illuminapairedend output file to discard sequences indicated as “joined”, so not assembled (“alignment”) (see explanation about this mode on the previous step)
Hands On: Remove unaligned sequence records
- obigrep ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
ilumimnapairedend fastq groomer output file- “Choose the sequence record selection option”:
predicat
- “Python boolean expression to be evaluated for each sequence record.”:
mode!="joined"
- You can use FastQC ( Galaxy version 0.73+galaxy0) to look at the format of the sequencing encoding score then FASTQ GROOMER ( Galaxy version 1.1.5) to specify the guessed sequencing encoding score and create a fastqsanger file.
CommentThe obigrep command is in some way analog to the standard Unix grep command. It selects a subset of sequence records from a sequence file.
A sequence record is a complex object composed of an identifier, a set of attributes (key=value), a definition, and the sequence itself.
Instead of working text line by text line as the standard Unix tool, selection is done sequence record by sequence record. A large set of options allows refining selection on any of the sequence record elements.
Moreover obigrep allows specifying simultaneously several conditions (that take the value TRUE or FALSE) and only the sequence records that fulfill all the conditions (all conditions are TRUE) are selected. You can refer to https://pythonhosted.org/OBITools/scripts/obigrep.html for more details
Question
- How do you verify the operation is successful?
- How many sequences are kept? Discarded?
- you can search in the input file content the presence of
mode=joinedand same on the output file (just clicking the eye to visualize the content of each file and typing CTRL+C for example to searchmode=joinedin the file, or using a regex Galaxy tool for example). You can also at least look at the size of the output file, if smaller than input file, this is a first good indication.- You can use a Galaxy tool like
Line/Word/Character count of a datasetto count the number of lines of each dataset (input and output of obigrep) and divided by 4 (as in a FastQ file, each sequence is represented by a block of 4 lines). 45 276 sequences for input file. 44 717 for output file. Thus 559 sequences discarded.
Assign each sequence record to the corresponding sample/marker combination with NGSfilter
Hands On: Assigns sequence records to the corresponding experiment/sample based on DNA tags and primersCommentEach sequence record is assigned to its corresponding sample and marker using the data provided in a text file (here wolf_diet_ngsfilter.txt). This text file contains one line per sample, with the name of the experiment (several experiments can be included in the same file), the name of the tags (for example: aattaac if the same tag has been used on each extremity of the PCR products, or aattaac:gaagtag if the tags were different), the sequence of the forward primer, the sequence of the reverse primer, the letter T or F for sample identification using the forward primer and tag only or using both primers and both tags, respectively.
- NGSfilter ( Galaxy version 1.2.13) with the following parameters:
- “Parameter file”:
wolf_diet_ngsfilter- “Read from file”:
obigrep output- “Number of errors allowed for matching primers”:
2- “Output data type”:
fastq
- If you are sure the format is compatible with a tabular datatype, as this is the case here ;), you can manually change it, clicking on the eye of the “wolf_diet_ngsfilter.txt” dataset, then selecting the “Datatypes” section then affecting manually tabular and saving the operation
Question
- How many sequences are not assigned?
- 1391
Dereplicate reads into uniq sequences with obiuniq
Hands On: Groups together sequence recordsCommentThe same DNA molecule can be sequenced several times. In order to reduce both file size and computations time, and to get easier interpretable results, it is convenient to work with unique sequences instead of reads. To dereplicate such reads into unique sequences, we use the obiuniq command. Definition: Dereplicate reads into unique sequences
- compare all the reads in a data set to each other
- group strictly identical reads together
- output the sequence for each group and its count in the original dataset (in this way, all duplicated reads are removed) Definition adapted from Seguritan and Rohwer (2001)
- obiuniq ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
Trimmed and annotated file by NGSfilter- “Attribute to merge”:
sample- “Use specific option”:
merge
Question
- How many sequences you had and how many you finally obtain?
- From 43 326 to 3 962
Limit number of informations with obiannotate
Hands On: Adds/Edits sequence record annotationsCommentobiannotate is the command that allows adding/modifying/removing annotation attributes attached to sequence records. Once such attributes are added, they can be used by the other OBITools commands for filtering purposes or for statistics computing.
Here, the goal is to keep only
countandmerged_samplekey=value attributes!
- obiannotate ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obiuniq output file- In “Keep only attribute with key”:
- “key”:
count- “if you want to specify a second key”:
merged_sample
Computes basic statistics for attribute values with obistat
Hands On: Computes basic statistics for attribute valuesCommentstats computes basic statistics for attribute values of sequence records. The sequence records can be categorized or not using one or several -c options. By default, only the number of sequence records and the total count are computed for each category. Additional statistics can be computed for attribute values in each category, like:
- minimum value (-m option)
- maximum value (-M option)
- mean value (-a option)
- variance (-v option)
- standard deviation (-s option)
The result is a contingency table with the different categories in rows, and the computed statistics in columns.
- obistat ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obiannotate output file- In “Category attribute”:
- param-repeat “Insert Category attribute”
- “How would you specify the category attribute key?”:
simply by a key of an attribute
- “Attribute used to categorize the sequence records”:
count- “Use a specific option”:
no
Question
- Can you use this result to say how many sequences occuring only once? You would need to use Galaxy tools like
Sort data in ascending or descending orderand ` Select first lines from a dataset` to answer the question
- 3131 sequences are occuring once.
Filtering sequances by count and length with obigrep
In this step, we are going to use obigrep in order to keep only the sequences having a count greater or equal to 10 and a length shorter than 80 bp.
Hands On: filter sequences with obigrep
- obigrep ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obiannotate output file- “Choose the sequence record selection option”:
predicat
- “Python boolean expression to be evaluated for each sequence record.”:
count>=10- obigrep ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obigrep output file- “Choose the sequence record selection option”:
lmin
- “lmin”:
80CommentBased on the previous observation, we set the cut-off for keeping sequences for further analysis to a count of 10 Based on previous knowledge we also remove sequences with a length shorter than 80 bp (option -l) as we know that the amplified 12S-V5 barcode for vertebrates must have a length around 100bp
Question
- How many sequences are kept following the “count” filter?
- How many sequences are kept following the “length” filter?
- 178
- 175
Clean the sequences for PCR/sequencing errors (sequence variants) with obiclean
Hands On: Clean the sequences for PCR/sequencing errors (sequence variants)As a final denoising step, using the obiclean program, we keep the head sequences that are sequences with no variants with a count greater than 5% of their own count
- obiclean ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obigrep output file- “attribute containing sample definition”:
merged_sample- “Maximum numbers of differences between two variant sequences (default: 1)”:
1- “Threshold ratio between counts (rare/abundant counts) of two sequence records so that the less abundant one is a variant of the more abundant (default: 1, i.e. all less abundant sequences are variants)”:
0.05- “Do you want to select only sequences with the head status in a least one sample?”:
Yes
Taxonomic assignment of sequences with NCBI BLAST+ blastn
Hands On: Search nucleotide database with nucleotide query sequence(s) from OBITools treatmentsOnce denoising has been done, the next step in diet analysis is to assign the barcodes to the corresponding species in order to get the complete list of species associated to each sample. Taxonomic assignment of sequences requires a reference database compiling all possible species to be identified in the sample. Assignment is then done based on sequence comparison between sample sequences and reference sequences. We here propose to use BLAST+ blastn.
- NCBI BLAST+ blastn ( Galaxy version 2.10.1+galaxy0) with the following parameters:
- “Nucleotide query sequence(s)”:
obiclean output file- “Subject database/sequences”:
FASTA file from your history- “Nucleotide FASTA subject file to use instead of a database”:
db_v05_r117- “Set expectation value cutoff”:
0.0001- “Output format”:
Tabular (extended 25 columns)- “Advanced Options”:
Show Advanced Options- *“Maximum hits to consider/show”:
1CommentHere we directly use the
db_v05_r117fasta file proposed on the original obitools tutorial. One can mention you can create such a fasta file using same obitools workflow describe before (using obigrep/obiuniq/obigrep/obiannotate) on downloaded EMBL datrabases and taxonomy treated by obitools ecoPCR tool.
Filter database and query sequences by ID to re associate informations with Filter sequences by ID
Hands On: Filter Blast resultsCommentThis tool allows you to re-associate all the reference sequences information, notably the
species_nameone so you can see which species are potentially seen on the sample. We will also use it to re-associate all the query sequences information, notably themerged_sampleandobiclean_countattributes so we can better evaluate quality of the results.
- Filter sequences by ID ( Galaxy version 0.2.7) with the following parameters:
- “Sequence file to be filtered”:
db_v05_r117- “Filter using the ID list from”:
tabular file- “Tabular file containing sequence identifiers”:
megablast on obiclean output- “Column(s) containing sequence identifiers”:
Column 2- “Output positive matches, negative matches, or both?”:
just positive match- Filter sequences by ID ( Galaxy version 0.2.7) with the following parameters:
- “Sequence file to be filtered”:
obiclean output data- “Filter using the ID list from”:
tabular file- “Tabular file containing sequence identifiers”:
megablast on obiclean output- “Column(s) containing sequence identifiers”:
Column 1- “Output positive matches, negative matches, or both?”:
just positive match
From FASTA to tabular with Obitab
Hands On: Convert fasta filtered files in tabular ones
- obitab ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
db_v05_r117 with matched IDCommentThis tool allows you to convert a fasta file into a tabular one so it is easier to read sequences definitions.
- obitab ( Galaxy version 1.2.13) with the following parameters:
- “Input sequences file”:
obiclean on data 61 with matched ID
create a final synthesis as a tabular file
Hands On: Join blast and obitab files then cut relevant column and apply filters
- Join two datasets side by side on a specified field with the following parameters:
- “Join”:
obitab on obiclean output file- “using column”:
Column 1- “with”:
megablast output file- “using column”:
Column 1- “Fill empty columns”:
Yes- “Fill Columns by”:
Single fill value- “Fill value”:
NA- Join two datasets side by side on a specified field with the following parameters:
- “Join”: last
Join two Datasets output file- “using column”:
Column 26- “with”:
obitab on db_v05_r117 with matched ID output file- “using column”:
Column 1- “Fill empty columns”:
Yes- “Fill Columns by”:
Single fill value- “Fill value”:
NACommentTo have something easier to read and understand, we create a tabular file containing only columns with important informations (c1: query sequences names / c3-7: query counts / c50: reference sequences names / c54: family / c59: genus / c51: reference annotations).
- Cut columns from a table with the following parameters:
- “Cut columns”:
c1,c3,c4,c5,c6,c7,c50,c54,c59,c51- “From”: last
Join two Datasets output file- Filter data on any column using simple expressions with the following parameters:
- “Filter”:
Cut output file- “With following condition”:
c3>1000 or c4>1000 or c5>1000 or c6>1000- “Number of header lines to skip”:
1CommentTo keep only data with significative counts.
Question
- How many species are identified? You can use
Cut columns from a tableandunique occurences of each recordto isolate thespecies namecolumn of obitab results.- Can you deduce the diet of each sample? You can use tools like
obitabandJoin two Datasets side by side on a specified fieldto join megablast results to obigrep one anddb_v05_r117 with matched ID
- 1O
- If we remove human sequences ;), some squirrel (sample:26a_F040644), deer (sample:15a_F730814 + sample:29a_F260619), stag (sample:29a_F260619 + sample:13a_F730603), marmot (sample:26a_F040644), doe (sample:29a_F260619), Grimm’s duiker (sample:29a_F260619).
Conclusion
You just did a ecological analysis, finding diet from wolves faeces ! So now you know how to preprocess metabarcoding data on Galaxy, producing quantitative informations with quality checks and filtering results to interpret it and to have a synthesis table you can share broadly!
You've Finished the Tutorial
Key points
From raw reads you can process, clean and filter data to obtain a list of species from environmental DNA (eDNA) samples.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Schloss, P. D., S. L. Westcott, T. Ryabin, J. R. Hall, M. Hartmann et al., 2009 Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75: 7537–7541.
- Caporaso, J. G., J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman et al., 2010 QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7: 335–336.
- Ficetola, G. F., E. Coissac, S. Zundel, T. Riaz, W. Shehzad et al., 2010 An In silico approach for the evaluation of DNA barcodes. BMC Genomics 39: e145–e145. 10.1186/1471-2164-11-434
- Riaz, T., W. Shehzad, A. Viari, F. Pompanon, P. Taberlet et al., 2011 ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Research 39: e145–e145. 10.1093/nar/gkr732
- SHEHZAD, W. A. S. I. M., T. I. A. Y. Y. B. A. RIAZ, M. U. H. A. M. M. A. D. A. NAWAZ, C. H. R. I. S. T. I. A. N. MIQUEL, C. A. R. O. L. E. POILLOT et al., 2012 Carnivore diet analysis based on next-generation sequencing: application to the leopard cat (Prionailurus bengalensis) in Pakistan. Molecular Ecology 21: 1951–1965. 10.1111/j.1365-294x.2011.05424.x
- Boyer, F., C. Mercier, A. Bonin, Y. L. Bras, P. Taberlet et al., 2015 obitools: aunix-inspired software package for DNA metabarcoding. Molecular Ecology Resources 16: 176–182. 10.1111/1755-0998.12428
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Coline Royaux, Olivier Norvez, Eric Coissac, Frédéric Boyer, Yvan Le Bras, Metabarcoding/eDNA through Obitools (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/ecology/tutorials/Obitools-metabarcoding/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{ecology-Obitools-metabarcoding, author = "Coline Royaux and Olivier Norvez and Eric Coissac and Frédéric Boyer and Yvan Le Bras", title = "Metabarcoding/eDNA through Obitools (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/ecology/tutorials/Obitools-metabarcoding/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/ecology/tutorials/Obitools-metabarcoding/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: fastq_groomer owner: devteam revisions: 47e5dbc3e790 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastqc owner: devteam revisions: 3d0c7bdf12f5 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: ncbi_blast_plus owner: devteam revisions: 5edc472ec434 tool_panel_section_label: NCBI Blast tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: unzip owner: imgteam revisions: 38eec75fbe9b tool_panel_section_label: Collection Operations tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_annotate owner: iuc revisions: d50dc96e3ce9 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_clean owner: iuc revisions: 816cbd8f4e6e tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_grep owner: iuc revisions: 02bab5ff7c37 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_illumina_pairend owner: iuc revisions: 8cb6bd511879 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_ngsfilter owner: iuc revisions: 91a1ed0b486f tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_stat owner: iuc revisions: 5f4544915893 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_tab owner: iuc revisions: 4cf47c504672 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: obi_uniq owner: iuc revisions: d4bea99366f9 tool_panel_section_label: DNA Metabarcoding tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: seq_filter_by_id owner: peterjc revisions: 141612f8c3e3 tool_panel_section_label: Filter and Sort tool_shed_url: https://toolshed.g2.bx.psu.edu/