Exome sequencing data analysis for diagnosing a genetic disease
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How do you identify genetic variants in samples based on exome sequencing data?
How do you, among the set of detected variants, identify candidate causative variants for a given phenotype/disease?
Requirements:
Jointly call variants and genotypes for a family trio from whole-exome sequencing data
Use variant annotation and the observed inheritance pattern of a phenotype to identify candidate causative variants and to prioritize them
- Introduction to Galaxy Analyses
- slides Slides: Quality Control
- tutorial Hands-on: Quality Control
- slides Slides: Mapping
- tutorial Hands-on: Mapping
Time estimation: 5 hoursSupporting Materials:Published: Aug 19, 2016Last modification: Mar 9, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00311rating Rating: 4.5 (2 recent ratings, 45 all time)version Revision: 30
Exome sequencing is a method that enables the selective sequencing of the exonic regions of a genome - that is the transcribed parts of the genome present in mature mRNA, including protein-coding sequences, but also untranslated regions (UTRs).
In humans, there are about 180,000 exons with a combined length of ~ 30 million base pairs (30 Mb). Thus, the exome represents only 1% of the human genome, but has been estimated to harbor up to 85% of all disease-causing variants (Choi et al., 2009).
Exome sequencing, thus, offers an affordable alternative to whole-genome sequencing in the diagnosis of genetic disease, while still covering far more potential disease-causing variant sites than genotyping arrays. This is of special relevance in the case of rare genetic diseases, for which the causative variants may occur at too low a frequency in the human population to be included on genotyping arrays.
Of note, a recent study focusing on the area of clinical pediatric neurology indicates that the costs of exome sequencing may actually not be higher even today than the costs of conventional genetic testing (Vissers et al., 2017).
In principle, the steps illustrated in this tutorial are suitable also for the analysis of whole-genome sequencing (WGS) data. At comparable mean coverage, however, WGS datasets will be much larger than exome sequencing ones and their analysis will take correspondingly more time.
The obvious benefit of WGS compared to exome-sequencing, of course, is that it will allow variant detection in even more regions of the genome. As a less apparent advantage, the more complete information of WGS data can make it easier to detect copy number variation (CNV) and structural variants such as translocations and inversions (although such detection will require more sophisticated analysis steps, which are not covered by this tutorial).
Very generally, one could argue that exome-sequencing captures most of the information that can be analyzed with standard bioinformatical tools today at reasonable costs. WGS, on the other hand, captures as much information as today’s sequencing technology can provide, and it may be possible to reanalyze such data with more powerful bioinformatical software in the future to exploit aspects of the information that were not amenable to analysis at the time of data acquisition.
The identification of causative variants underlying any particular genetic disease is, as we will see in this tutorial, not just dependent on the successful detection of variants in the genome of the patient, but also on variant comparison between the patient and selected relatives. Most often family trio data, consisting of the genome sequences of the patient and their parents, is used for this purpose. With multisample data like this it becomes possible to search for variants following any kind of Mendelian inheritance scheme compatible with the observed inheritance pattern of the disease, or to detect possibly causative de-novo mutations or loss-of-heterozygosity (LOH) events.
This tutorial focuses on the practical aspects of analyzing real-world patient data. If you are more interested in the theoretical aspects of variant calling, you may want to have a look at the related tutorial on Calling variants in diploid systems.
The tutorial on Somatic variant calling follows an analysis workflow that is rather similar to the one here, but tries to identify tumor variants by comparing a tumor sample to healthy tissue from the same patient. Together, the two tutorials are intended to get you started with genomics medicine using Galaxy.
AgendaIn this tutorial, we will cover:
Data Preparation
In this tutorial, we are going to analyze exome sequencing data from a family trio, in which the boy child is affected by the disease osteopetrosis, while both parents, who happen to be consanguineous, are unaffected. Our goal is to identify the genetic variation that is responsible for the disease.
Get data
tip This tutorial offers two alternative entry points allowing you to
- conduct a full analysis starting from original sequenced reads in
fastqformat or to - start your analysis with premapped reads in
bamformat that are (almost) ready for variant calling.
While the full analysis is probably closer to how you would analyze your own data, the shortened analysis from premapped reads may suit your time frame better, and will avoid redundancy if you have previously worked through other tutorials demonstrating NGS quality control and read mapping, like the dedicated Quality control and Mapping tutorials.
The following hands-on section will guide you through obtaining the right data for either analysis.
Hands On: Data upload
Create a new history for this tutorial and give it a meaningful name
To create a new history simply click the new-history icon at the top of the history panel:
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter
Obtain the raw sequencing data
Comment: Starting from raw sequencing dataIn this and the following steps you will obtain the original unmapped sequencing data and prepare for a full analysis including the mapping of the sequenced reads.
If you prefer to skip the mapping step and start the analysis from premapped data you should proceed directly to step 4 of this section.
Import the original sequenced reads datasets of the family trio from Zenodo:
https://zenodo.org/record/3243160/files/father_R1.fq.gz https://zenodo.org/record/3243160/files/father_R2.fq.gz https://zenodo.org/record/3243160/files/mother_R1.fq.gz https://zenodo.org/record/3243160/files/mother_R2.fq.gz https://zenodo.org/record/3243160/files/proband_R1.fq.gz https://zenodo.org/record/3243160/files/proband_R2.fq.gzAlternatively, the same files may be available on your Galaxy server through a shared data library (your instructor may tell you so), in which case you may prefer to import the data directly from there.
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Change Type (set all): from “Auto-detect” to
fastqsanger.gzPress Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
Check that the newly created datasets in your history have their datatypes assigned correctly to
fastqsanger.gz, and fix any missing or wrong datatype assignment
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
fastqsanger.gzfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
trophy Congratulations for obtaining the datasets required for an analysis including reads mapping. You should now proceed with Step 7 below.
- Obtain the premapped sequencing data
Comment: Alternative entry point: Premapped dataSkip this and the following two steps if you already obtained and prepared the original unmapped seuencing data and are planning to perform the mapping step yourself.
Import the premapped reads datasets of the family trio from Zenodo:
https://zenodo.org/record/3243160/files/mapped_reads_father.bam https://zenodo.org/record/3243160/files/mapped_reads_mother.bam https://zenodo.org/record/3243160/files/mapped_reads_proband.bamAlternatively, the same files may be available on your Galaxy server through a shared data library (your instructor may tell you so), in which case you may prefer to import the data directly from there.
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Change Type (set all): from “Auto-detect” to
bamPress Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
Check that the newly created datasets in your history have their datatypes assigned correctly to
bam, and fix any missing or wrong datatype assignment
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
bamfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
Specify the genome version that was used for mapping
Change the database/build (dbkey) for each of your bam datasets to
hg19.
- Click the desired dataset’s name to expand it.
Click on the “?” next to database indicator:
- In the central panel, change the Database/Build field
- Select your desired database key from the dropdown list:
Human Feb. 2009 (GRCh37/hg19) (hg19)- Click the Save button
When you are starting with sequencing data that has already been mapped to a particular genome version (human hg19 in this case), it is good practice to attach this information as metadata to the datasets.
Doing so helps prevent accidental use of a different version of the reference genome in later steps like variant calling, which would typically lead to nonsensical results because of base position changes between the different versions.
trophy Congratulations for obtaining the premapped sequencing datasets. Now, follow the remaining steps to set everything up for a successful analysis.
Rename the datasets
For datasets that you upload via a link, Galaxy will pick the link address as the dataset name, which you will likely want to shorten to just the file names.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, change the Name field
- Click the Save button
Add #father/#mother/#child tags to the datasets
Parts of the analysis in this tutorial will consist of identical steps performed on the data of each family member.
To make it easier to keep track of which dataset represents which step in the analysis of which sample, Galaxy supports dataset tags. In particular, if you attach a tag starting with
#to any dataset, that tag will automatically propagate to any new dataset derived from the tagged dataset.Tags are supposed to help you identify the origin of datasets quickly, but you can choose them as you like.
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
- Click on the dataset to expand it
- Click on Add Tags galaxy-tags
- Add tag text. Tags starting with
#will be automatically propagated to the outputs of tools using this dataset (see below).- Press Enter
- Check that the tag appears below the dataset name
Tags beginning with
#are special!They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
- a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
- dataset 3 is used to calculate read coverage using BedTools Genome Coverage separately for
+and-strands. This generates two datasets (4 and 5 for plus and minus, respectively);- datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
- datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with
#plusand#minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.More information is in a dedicated #nametag tutorial.
Obtain the reference genome
Comment: ShortcutYou can skip this step if the Galaxy server you are working on offers a
hg19version of the human reference genome with prebuilt indexes for bwa-mem (only necessary if starting from unmapped original sequencing reads) and freebayes. Ask your instructor, or check the tools Map with BWA-MEM tool and FreeBayes tool if they list ahg19version as an option under “Select a / Using reference genome”).Import the
hg19version of the human chromosome 8 sequence:https://zenodo.org/record/3243160/files/hg19_chr8.fa.gzIn the upload dialog, make sure you specify:
- Type:
fasta- Genome:
Human Feb. 2009 (GRCh37/hg19) (hg19)Alternatively, load the dataset from a shared data library.
Rename the reference genome
The reference genome you have imported above came as a compressed file, but got unpacked by Galaxy to plain
fastaformat according to your datatype selection. At a minimum, you may now wish to remove the.gzsuffix from the dataset name to avoid confusion.



trophy Congratulations! You are all set for starting the analysis now.
If you have chosen to follow the complete analysis from the original sequenced data, just proceed with the next section.
If, on the other hand, you have prepared to start from the premapped data, skip the sections on Quality control and Read mapping, and conitnue with Mapped reads postprocessing.
Quality control
This step serves the purpose of identifying possible issues with the raw sequenced reads input data before embarking on any “real” analysis steps.
Some of the typical problems with NGS data can be mitigated by preprocessing affected sequencing reads before trying to map them to the reference genome. Detecting some other, more severe problems early on may at least save you a lot of time spent on analyzing low-quality data that is not worth the effort.
Here, we will perform a standard quality check on our input data and only point out a few interesting aspects about that data. For a more thorough explanation of NGS data quality control, you may want to have a look at the dedicated tutorial on Quality control.
To perform quality control, we will use Falco an efficiency-optimized rewrite of FastQC.
Hands On: Quality control of the input datasets
- Run Falco ( Galaxy version 1.2.4+galaxy0) on each of your six fastq datasets
- param-files “Short read data from your current history”: all 6 FASTQ datasets selected with Multiple datasets
- Click on param-files Multiple datasets
- Select several files by keeping the Ctrl (or COMMAND) key pressed and clicking on the files of interest
When you start this job, twelve new datasets (one with the calculated raw data, another one with an html report of the findings for each input dataset) will get added to your history.
- Use MultiQC ( Galaxy version 1.33+galaxy0) to aggregate the raw Falco data of all input datasets into one report
- In “Results”
“Which tool was used generate logs?”:
FastQCSince Falco produces FastQC-style output, MultiQC will work just fine with it and can parse it as if it had been generated with FastQC.
In “FastQC output”
- “Type of FastQC output?”:
Raw data- param-files “FastQC output”: all six RawData outputs of Falco tool)
Inspect the Webpage output produced by the tool
Question
- Based on the report, do you think preprocessing of the reads (trimming and/or filtering) will be necessary before mapping?
- Why do all samples show a non-normal GC content distribution, and should you be worried?
- Sequence quality is quite good overall. If anything you might consider trimming the 3’ ends of reads (base qualities decline slightly towards the 3’ ends) or to filter out the small fraction of reads with a mean base quality < 5. Feel free to run, e.g., Trimmomatic ( Galaxy version 0.39+galaxy2) on the fastq datasets if you want to, but don’t expect this to have a big effect on the analysis given the high overall quality of the data of all samples.
In whole-genome sequencing, a non-normal distribution of the GC content of the reads from a sample is typically considered to hint at possible contamination. Here, however, we are dealing with sequencing data from captured exomes, i.e, the reads are not representing random sequences from a genome, but rather a biased selection.
A bimodal GC content distribution, like for the samples at hand, is a characteristic feature of many exome capture methods and has also been observed with an Illumina Nextera Rapid Capture exome kit before (compare Fig. 3D in Shigemizu et al., 2015 ).
Read mapping
Now that you confirmed that the quality of the input data is good enough to warrant further analysis, it is time to map the sequenced reads to the reference genome.
We assume here that you are at least vaguely familiar with the concept of read mapping and only illustrate the concrete steps necessary to map our specific NGS reads datasets to the human reference genome. We recommend you to follow the dedicated Mapping tutorial, if you need a general introduction to read mapping.
Hands On: Read Mapping
- Map with BWA-MEM ( Galaxy version 0.7.19) to map the reads from the father sample to the reference genome
- “Will you select a reference genome from your history or use a built-in index?”:
Use a built-in genome index
- “Using reference genome”:
Human: hg19(or a similarly named option)Comment: Using the imported `hg19` sequenceIf you have imported the
hg19chr8 sequence as a fasta dataset into your history instead:
- “Will you select a reference genome from your history or use a built-in index?”:
Use a genome from history and build index
- param-file “Use the following dataset as the reference sequence”: your imported
hg19fasta dataset.- “Single or Paired-end reads”:
Paired
- param-file “Select first set of reads”: the forward reads (R1) dataset of the father sample
- param-file “Select second set of reads”: the reverse reads (R2) dataset of the father sample
Please confirm that the problematic datasets declare format:
fastqsanger.gz.Most Galaxy tools that accept FASTQ input expect the data to be formatted as FASTQ with Sanger-scaled quality values, the most widely spread version of the FASTQ format. To make this requirement explicit (instead of generating possibly wrong results) these tools require you to set the dataset type to
fastqsanger(fastqsanger.gzfor data compressed with gzip). You can do so either on data upload or later from the Edit dataset attributes view (which you can reach by clicking on the galaxy-pencil pencil icon.- “Set read groups information?”:
Set read groups (SAM/BAM specification)
- “Auto-assign”:
No
- “Read group identifier (ID)”:
000- “Auto-assign”:
No
- “Read group sample name (SM)”:
fatherWarning: Read group IDs and sample names - choose, but choose wiselyIn general, you are free to choose ID and SM values to your liking, but …
The ID should unambiguously identify the sequencing run that produced the reads. At the very least, no two input datasets in any given analysis should define the same ID twice, or tools like FreeBayes, which we are going to use in the next step, will refuse to work with the data.
The SM value, on the other hand, should identify the biological sample represented by the data and is used by many tools (like GEMINI which we will use later) to let you refer to one specifc sample in a multisample analysis. Choose descriptive, but short and easy to remember sample names since you will have to type them in again!
Map with BWA-MEM ( Galaxy version 0.7.19) to map the reads from the mother sample to the reference genome using the same parameters as before except
- “Single or Paired-end reads”:
Paired
- param-file “Select first set of reads”: the forward reads (R1) dataset of the mother sample
- param-file “Select second set of reads”: the reverse reads (R2) dataset of the mother sample
- “Set read groups information?”:
Set read groups (SAM/BAM specification)
- “Auto-assign”:
No
- “Read group identifier (ID)”:
001- “Auto-assign”:
No
- “Read group sample name (SM)”:
motherMap with BWA-MEM ( Galaxy version 0.7.19) to map the reads from the child sample to the reference genome using the same parameters as before except
- “Single or Paired-end reads”:
Paired
- param-file “Select first set of reads”: the forward reads (R1) dataset of the child sample
- param-file “Select second set of reads”: the reverse reads (R2) dataset of the child sample
- “Set read groups information?”:
Set read groups (SAM/BAM specification)
- “Auto-assign”:
No
- “Read group identifier (ID)”:
002- “Auto-assign”:
No
- “Read group sample name (SM)”:
proband
Mapped reads postprocessing
At this point in the analysis you should have obtained three mapped reads
datasets in bam format. Each of these datasets should:
-
have its database set to the key
hg19Please correct any missing (
?) or wrong keys now!- Click the desired dataset’s name to expand it.
-
Click on the “?” next to database indicator:

- In the central panel, change the Database/Build field
- Select your desired database key from the dropdown list:
Human Feb. 2009 (GRCh37/hg19) (hg19) - Click the Save button
-
ideally, carry one of the
#father,#motheror#childtags for quick identification of the samples they provide data for.
In principle, you could use these datasets directly for variant calling, and in many cases, including this one, this would be sufficient to identify the sought-after variants.
To obtain an accurate picture of the variant spectrum found in your samples it is good practice though to perform various postprocessing steps on the mapped reads before passing them to a variant caller.
Comment: ShortcutIf you are in a hurry, you may skip the postprocessing steps and continue with your mapped reads datasets and the Variant Calling section. Make sure though you come back here, or read about mapped reads postprocessing elsewhere, before attempting to perform any variant analysis on your own data that may require high accuracy/sensitivity.
The optimal set of postprocessing steps required depends on the variant calling software used at the next step. The FreeBayes variant caller that we are going to use in this tutorial is particularly well suited for use with minimal mapped reads postprocessing pipelines, so all we are going to do here is:
-
filter the paired-end reads of all samples to retain only those read pairs, for which both the forward and the reverse read have been mapped to the reference successfully
For such pairs of reads, we can be extra confident that they don’t come from some non-human contaminant DNA or represent a sequencing artefact of some sort.
-
deduplicate reads
Duplicate reads, which typically arise from PCR-overamplification of genomic fragments during sequencing library preparation, can, to some extent, lead to wrong genotype assignments at variant sites (if, for example, a sample is heterozygous for a variant, but fragments with one of the two alleles get amplified more efficiently than the others).
Comment: More postprocessing stepsThe Somatic variant calling tutorial provides an example of a much more complex mapped reads postprocessing pipeline that may be required for optimal performance of other variant callers.
Filtering on mapped reads properties
To produce new filtered BAM datasets with only mapped reads the mate of which is also mapped:
Hands On: Filtering for read pair mapping status
- Samtools view ( Galaxy version 1.22+galaxy1) with the following parameters (leaving non-mentioned ones at their defaults):
- param-files “SAM/BAM/CRAM data set”: all 3 mapped reads datasets of the family trio, outputs of Map with BWA-MEM tool
- “What would you like to look at?”:
A filtered/subsampled selection of reads
- In “Configure filters”:
- “Exclude reads with any of the following flags set”:
Read is unmappedandMate is unmapped
This will result in three new datasets, one for each sample in the analysis.
Instead of the above filter conditions we could also have exploited the Read is mapped in a proper pair flag bit.
For a read to be flagged as being mapped in a proper pair its mate needs to be mapped, but the mapped pair also needs to meet additional, aligner-specific criteria. These may include (and do so for BWA-MEM):
- both reads need to map to the same chomosome
- the two reads need to map to the reference in the orientation expected by the aligner
- the two read pairs need to map to the reference within an aligner-determined distance
Thus, filtering based on the flag has two consequences:
- filtering will be stricter than with just the Read is unmapped and Mate is unmapped flags above
you will eliminate read pairs that could be informative with regard to chromosomal rearrangements and insertion/deletion events
=> Do not filter for properly paired reads if you plan to detect such structural variants!
In addition, the proper pair flag is considered undefined if the read itself is unmapped, so a proper pair filter should eliminate unmapped reads explicitly to be on the safe side.
Thus, if you would like to use proper pair filtering (we have no intention to detect structural variants in this tutorial) instead of just filtering for mapped reads with a mapped mate, you could run the alternative:
Hands On
- Samtools view ( Galaxy version 1.22+galaxy1):
- param-files “SAM/BAM/CRAM data set”: all 3 mapped reads datasets of the family trio, outputs of Map with BWA-MEM tool
- “What would you like to look at?”:
A filtered/subsampled selection of reads
- In “Configure filters”:
- “Require that these flags are set”:
Read is mapped in a proper pair- “Exclude reads with any of the following flags set”:
Read is unmapped
Removing duplicate reads
Hands On: Remove duplicates
- RmDup ( Galaxy version 2.0.1) with the following parameters:
- param-files “BAM file”: all 3 filtered reads datasets; the outputs of Samtools view
- “Is this paired-end or single end data”:
BAM is paired-end
- “Treat as single-end”:
No
Again, this will produce three new datasets, one for each member of the family trio.
Variant calling
With the sequenced reads of all samples mapped and postprocessed, we can start looking for evidence of sequence deviations, i.e. variants, between the sequenced genomic samples and the reference genome.
This task has been automated and optimized continuously over the last decade, and modern variant calling software hides much of the complexity involved in it. At least a basic understanding of the underlying concepts is still highly recommended though and, if you are new to variant calling, the tutorial on Calling variants in diploid systems may be a good starting point for you.
Generating FreeBayes calls
We will use FreeBayes to call our variants. FreeBayes is a Bayesian genetic variant detector designed to find small polymorphisms, specifically SNPs (single-nucleotide polymorphisms), indels (insertions and deletions), MNPs (multi-nucleotide polymorphisms), and complex events (composite insertion and substitution events) smaller than the length of a short-read sequencing alignment.
Hands On: Generating FreeBayes calls
- Run FreeBayes ( Galaxy version 1.3.10+galaxy1):
- “Choose the source for the reference genome”:
Locally cached
- “Run in batch mode?”:
Merge output VCFs
- param-files “BAM dataset(s)”: all three mapped reads datasets of the family trio; the outputs of RmDup
- “Using reference genome”:
Human: hg19(or a similarly named option)Comment: Using the imported `hg19` sequenceIf you have imported the
hg19chr8 sequence as a fasta dataset into your history instead:
- “Choose the source for the reference genome”:
History
- “Run in batch mode?”:
Merge output VCFs
- param-files “BAM or CRAM dataset(s)”: all three mapped and fully post-processed reads datasets of the family trio; outputs of RmDup
- param-file “Use the following dataset as the reference sequence”: your imported
hg19fasta dataset.- “Limit variant calling to a set of regions?”:
Do not limit- “Choose parameter selection level”:
1. Simple diploid calling
trophy Congratulations! You have created you first multisample VCF file, one of the most complicated file formats in bioinformatics. For every variant detected in at least one of your samples, this tab-separated format uses a single line to store all information about the variant. This includes but is not limited to:
- the position of the variant in the genome (with respect to the reference genome used for the analysis)
- the nature of the variant (the actual sequence change associated with it)
- the detected genotype of every sample at the variant position
- measures of the reliability of the variant call and of all individual genotype calls
Hands On: Optional hands-on: Inspect the VCF output produced by FreeBayes
- Display the VCF dataset:
Click the galaxy-eye icon next to the VCF dataset generated by FreeBayes to display its contents.
VCF is a tabular plain text format though its information density makes it complicated to understand.
QuestionCan you locate at least some of the above-listed information in the dataset?
Hints:
- Lines starting with
##are comment lines explaining the content of the file.- Diploid genotypes at biallelic sites are encoded using
0/0,0/1and1/1to represent homozygous reference, heterozygous and homozygous variant states, respectively.
The position of each variant with respect to the reference genome is stored in the first two columns (labeled
CHROMandPOS).The sequence change associated with the variant is provided by columns 4 and 5 (
REFandALT).The first of these lists the sequence found at the variant site in the reference genome, the second the variant sequence.
The genotypes of the samples are stored as part of columns 10 and up.
Column 9 (
FORMAT) provides the legend for understanding the sample-specific columns following it and tells us that the first element (before the first:separator) of each sample column holds the sample genotype (GT).The variant call quality (a measure of the reliability of the variant existing in any of the samples, i.e. it being real) is stored in column 6 (
QUAL). The values are Phred-scaled like sequenced reads base qualities.The (log10-scaled) likelihoods of individual genotype calls are stored as the last element (
GL) in every sample column.If you are eager to learn all the details about the format, you could consult the excellent (but also long) official VCF specification.
Post-processing FreeBayes calls
Before starting to analyze the detected variants, we need to post-process the VCF dataset to fix a few incompatibilities between Freebayes and downstream analysis tools. In particular, we want to:
-
Split multiallelic variant records, i.e., records that list more than one alternate allele at a given genomic position, into separate record lines.
This will allow us to annotate each record with information about the impact of one specific variant allele further on.
-
Make sure that indels are represented in left-aligned and normalized form because this is how known indels are stored in public annotation databases.
Indel normalization is a surprisingly complex topic, which is explained really well and in detail in Tan et al., 2015. The following figure illustrates the various possibilities to represent an indel, only one of which is the normalized form:
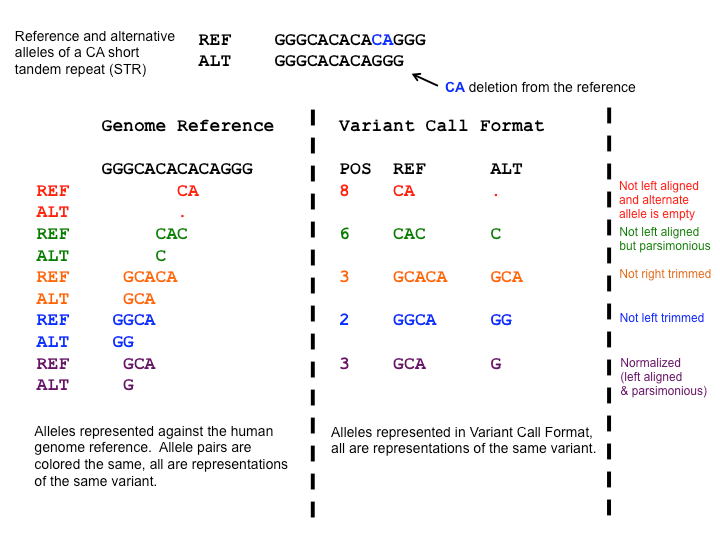 Open image in new tab
Open image in new tabFigure 1: The many ways to represent an indel (from: https://genome.sph.umich.edu/wiki/Variant_Normalization .
A tool that can do this and also ensures that a VCF dataset conforms to standards in some other, less important respects is bcftools norm.
Hands On: Post-processing FreeBayes calls
- bcftools norm ( Galaxy version 1.22+galaxy0) with the following parameters:
- “VCF/BCF Data”: the VCF output of FreeBayes tool
- “Choose the source for the reference genome”:
Use a built-in genome
- “Reference genome”:
Human: hg19(or a similarly named option)Comment: Using the imported `hg19` sequenceIf you have imported the
hg19chr8 sequence as a fasta dataset into your history instead:
- “Choose the source for the reference genome”:
Use a genome from the history
- “Reference genome”: your imported
hg19fasta dataset- “When any REF allele does not match the reference genome base”:
ignore the problem (-w)- “Atomize”:
Do not atomize- “Left-align and normalize indels?”:
Yes“Perform deduplication for the folowing types of variant records”:
do not deduplicate any recordsFreebayes is not producing any duplicate calls.
- “~multiallelics”:
split multiallelic sites into biallelic records (-)
“split the following variant types”:
bothWe want to split both, multiallelic SNP and indel records.
“output_type”:
uncompressed VCFWe would like to keep the results human-readable. VCF is also what tools like SnpEff and GEMINI expect as input. Compressed, binary BCF is interesting for space-efficient long-term storage of large lists of variants.
You could try to look for the differences between the original and the normalized VCF dataset, but for convenience bcftools norm reports a brief summary of the actions it performed. Expand the dataset in the history (by clicking on its name) to see this output listing the total number of variant lines processed, along with the number of split, realigned and skipped records.
Variant annotation and reporting
A list of variants detected in a set of samples is a start, but to discover biologically or clinically relevant information in it is almost impossible without some additional tools and data. In particular, the variants in the list need to be:
-
prioritized with respect to their potential relevance for the biological / clinical phenotype that is studied
Even with exome sequencing, only a fraction of the detected variants will have a clear impact on the function of a protein (many variants will introduce silent mutations, or reside in intronic regions still covered by the exome-enriched sequencing data). Of these, many will have been observed before in healthy individuals arguing against them playing an important role in an adverse phenotype.
-
filtered based on the inheritance pattern expected for a causative variant
A multisample VCF file records the most likely genotypes of all samples at every variant site. Knowing which individuals (samples) are affected by a phenotype we can exclude variants with inheritance patterns that are incompatible with the observed inheritance of the phenotype.
-
reported in a more human-friendly form
While the VCF format can be used to encode all relevant information about any variant, it is hard for humans to parse that information.
Ideally, one would like to generate simpler reports for any set of filtered and prioritized variants.
Get data
Our workhorse for annotating and reporting variants and the genes affected by them will be the GEMINI framework. GEMINI comes bundled with a wealth of annotation data for human variants from many different sources. These can be used to annotate any list of human variants conveniently, without the need for separate downloads and conversion between different annotation data formats.
Beyond its bundled annotation data, GEMINI also provides (limited) support for using custom annotations. The Somatic variant calling tutorial demonstrates the use of GEMINI annotate tool for this purpose.
The only additional annotation tool we need, for the purpose of this tutorial, is the tool SnpEff, which can annotate variants with their calculated effects on known genomic features. Because SnpEff is a generic tool that can be used on variants found in the genome of any organism we need to provide it with a so-called SnpEff genome file that holds the annotated features (genes, transcripts, translated regions, etc.) for our genome of interest.
While annotated variants are all we need to prioritize them as described above, filtering based on inheritance patterns requires a way to inform GEMINI about the relationship between our samples and their observed phenotypes. This is done through a so-called pedigree file in PED format, which is rather simple to generate manually.
Hands On: Obtain SnpEff genome and GEMINI pedigree files
Download SnpEff functional genomic annotations
Comment: ShortcutYou can skip this step if the Galaxy server you are working on offers
Homo sapiens: hg19as a locally installed snpEff database. You can check the Genome source select list of the SnpEff eff ( Galaxy version 5.2+galaxy1) tool to see if this is the case.Use SnpEff Download ( Galaxy version 5.2+galaxy1) to download genome annotation database
hg19.Create a PED-formatted pedigree dataset describing our single-family sample trio:
#family_id name paternal_id maternal_id sex phenotype FAM father 0 0 1 1 FAM mother 0 0 2 1 FAM proband father mother 1 2and set its datatype to
tabular.
- Click galaxy-upload Upload Data at the top of the tool panel
- Select galaxy-wf-edit Paste/Fetch Data at the bottom
- Paste the file contents into the text field
- Change Type from “Auto-detect” to
tabular* From the Settings menu (galaxy-gear) select Convert spaces to tabs* Press Start and Close the windowWarning: Remember those sample namesThe above content of the pedigree dataset assumes you chose
father,mother,probandas the sample names at the read mapping step (if you haven’t mapped the reads yourself, but started with the premapped data, you can safely skip this warning section). By now, these sample names will have been propagated through BWA-MEM and Freebayes to the VCF dataset of variants. It is important that you use matching sample names in the pedigree and in the VCF dataset, or GEMINI will not be able to connect the information in them.If you have chosen different sample names before, you have to adjust the pedigree dataset accordingly!
The PED format is explained in the help section of GEMINI load ( Galaxy version 0.20.1+galaxy2).
Take a moment and try to understand the information that is encoded in the PED dataset we are using here.
Variant annotation with functional genomic effects
We need to start annotating our variants with SnpEff simply because Gemini knows how to parse SnpEff-annotated VCFs, while GEMINI output cannot be used with SnpEff.
Hands On: Adding annotations with SnpEff
- SnpEff eff ( Galaxy version 5.2+galaxy1)
- param-file “Sequence changes (SNPs, MNPs, InDels)”: the output of bcftools norm tool
- “Input format”:
VCF- “Output format”:
VCF (only if input is VCF)- “Produce Summary Stats”:
Yes- “Genome source”:
Locally installed snpEff database
- “Genome”:
Homo sapiens: hg19(or a similarly named option)Comment: Using the imported `hg19` SnpEff genome databaseIf you have imported the
hg19SnpEff genome database into your history instead:
- “Genome source”:
Downloaded snpEff database in your history
- param-file “SnpEff4.3 Genome Data”: your imported
hg19SnpEff dataset.
Running the above job will produce two datasets. One is a Summary Stats HTML report, which contains some interesting general metrics such as a distribution of variants across gene features. The other one is the main annotation result - a VCF like the input, but with annotations of variant effects added to the INFO column.
Hands On: Optional hands-on: Inspect the Summary Stats output produced by SnpEff
- Display the dataset:
- Click the galaxy-eye icon next to the HTML dataset generated by SnpEff to display its contents.
QuestionOne section in the report is Number of effects by type and region. Given that you are analyzing exome data, what is the most surprising aspect in this section? Do you have an idea how to explain it?
According to the report, intronic variants make up 50% of all variants detected!
Exome capture kits are designed to capture exons plus a bit of surrounding sequence to ensure proper coverage of the exon ends including splice junction sites.
Thus, even though intronic sequences are underrepresented in exome sequencing data, not all of them are eliminated. Since mutations in most intron bases are neutral, they can accumulate at higher frequency than most mutations in exons and, thus, still represent a relevant fraction of all detected variants.
Generating a GEMINI database of variants for further annotation and efficient variant queries
Next, we are going to use the SnpEff-annotated VCF as the basis for more exhaustive annotation with GEMINI. Unlike SnpEff, GEMINI does not just add annotations to a list of variants in VCF format. Instead the framework extracts the variants from the VCF input and stores them, together with newly added annotations, in an SQL database. It then lets you formulate queries for retrieving and reporting subsets of variants. The combined variant extraction/annotation/storage step is performed by the GEMINI load tool. In addition, that same tool can be used to incorporate sample pedigree info into the database.
Hands On: Creating a GEMINI database from a variants dataset
- GEMINI load ( Galaxy version 0.20.1+galaxy2) with
- param-file “VCF dataset to be loaded in the GEMINI database”: the output of SnpEff eff tool
- “The variants in this input are”:
annotated with snpEff“This input comes with genotype calls for its samples”:
YesSample genotypes were called by Freebayes for us.
- “Choose a gemini annotation source”: select the latest available annotations snapshot (most likely, there will be only one)
- “Sample and family information in PED format”: the pedigree file prepared above
- “Load the following optional content into the database”
- param-check “GERP scores”
- param-check “CADD scores”
- param-check “Gene tables”
- param-check “Sample genotypes”
- param-check “variant INFO field”
Leave unchecked the following:
“Genotype likelihoods (sample PLs)”
Freebayes does not generate these values
“only variants that passed all filters”
This setting is irrelevant for our input because Freebayes did not apply any variant filters.
Running this job generates a GEMINI-specific database dataset, which can only be processed with other GEMINI tools. The benefit, however, is that we now have variants, rich annotations and pedigree info stored in a format that enables flexible and highly efficient queries, which will greatly simplify our actual task to identify the variant responsible for the child’s disease!
For a beginner, the sheer number of GEMINI tools may be a bit daunting and give the impression that this framework adds a lot of complexity. In practice, however, you will likely only need a very limited number of these tools for any given analysis. To help you get an overview, here is a list of the most general-purpose tools and their function:
- Every analysis will always start with GEMINI load tool to build the GEMINI database from a variant list in VCF format and the default annotations bundled with GEMINI.
- GEMINI amend tool and GEMINI annotate tool let you add additional information to a GEMINI database after it has been created. Use GEMINI amend to overwrite the pedigree info in a database with the one found in a given PED input, and GEMINI annotate to add custom annotations based on a VCF or BED dataset.
- GEMINI database info tool lets you peek into the content of a GEMINI database to explore, e.g, what annotations are stored in it.
- GEMINI query tool and GEMINI gene-wise tool are the two generic and most flexible tools for querying, filtering and reporting variants in a database. There is hardly anything you cannot do with these tools, but you need to know or learn at least some SQL-like syntax to use them.
- GEMINI inheritance pattern tool simplifies variant queries based on their inheritance pattern. While you could use the query and gene-wise tools for the same purpose, this tool allows you to perform a range of standard queries without typing any SQL. This tool is what we are going to use here to identify the causative variant.
- The remaining tools serve more specialized purposes, which are beyond the scope of this tutorial.
The Somatic variant calling tutorial demonstrates the use of the GEMINI annotate and GEMINI query tools, and tries to introduce some essential bits of GEMINI’s SQL-like syntax.
For a thorough explanation of all tools and functionality you should consult the GEMINI documentation.
Candidate variant detection
Let us now try to identify variants that have the potential to explain the boy child’s osteopetrosis phenotype. Remember that the parents are consanguineous, but both of them do not suffer from the disease. This information allows us to make some assumptions about the inheritance pattern of the causative variant.
QuestionWhich inheritance patterns are in line with the phenotypic observations for the family trio?
Hint: GEMINI easily lets you search for variants fitting any of the following inheritance patterns:
- Autosomal recessive
- Autosomal dominant
- X-linked recessive
- X-linked dominant
- Autosomal de-novo
- X-linked de-novo
- Compound heterozygous
- Loss of heterozygosity (LOH) events
Think about which of these might apply to the causative variant.
- Since both parents are unaffected the variant cannot be dominant and inherited.
- A de-novo acquisition of a dominant (or an X-linked recessive) mutation is, of course, possible.
- A recessive variant is a possibility, and a more likely one given the parents’ consanguinity.
- For both the de-novo and the inherited recessive case, the variant could reside on an autosome or on the X chromosome. Given that we provided you with only the subset of sequencing reads mapping to chr8, an X-linked variant would not be exactly instructive though ;)
- A compound heterozygous combination of variant alleles affecting the same gene is possible, but less likely given the consanguinity of the parents (as this would require two deleterious variant alleles in the gene circulating in the same family).
- A loss of heterozygosity (LOH) turning a heterozygous recessive variant into a homozygous one could be caused by uniparental disomy or by an LOH event early in embryonic development, but both these possibilities have an exceedingly small probability.
Based on these considerations it makes sense to start looking for inherited autosomal recessive variants first. Then, if there is no convincing candidate mutation among them, you could extend the search to de-novo variants, compund heterozygous variant pairs and LOH events - probably in that order.
Since our GEMINI database holds the variant and genotype calls for the family trio and the relationship between the family members, we can make use of GEMINI inheritance pattern tool to report all variants fitting any specific inheritance model with ease.
Below is how you can perform the query for inherited autosomal recessive variants. Feel free to run analogous queries for other types of variants that you think could plausibly be causative for the child’s disease.
Hands On: Finding and reporting plausible causative variants
- GEMINI inheritance pattern ( Galaxy version 0.20.1)
- “GEMINI database”: the GEMINI database of annotated variants; output of GEMINI load tool
- “Your assumption about the inheritance pattern of the phenotype of interest”:
Autosomal recessive
- param-repeat “Additional constraints on variants”
“Additional constraints expressed in SQL syntax”:
impact_severity != 'LOW'This is a simple way to prioritize variants based on their functional genomic impact. Variants with low impact severity would be those with no obvious impact on protein function (i.e., silent mutations and variants outside coding regions)
“Include hits with less convincing inheritance patterns”:
NoThis option is only meaningful with larger family trees to account for errors in phenotype assessment.
“Report candidates shared by unaffected samples”:
NoThis option is only meaningful with larger family trees to account for alleles with partial phenotypic penetrance.
“Family-wise criteria for variant selection”: keep default settings
This section is not useful when you have data from just one family.
- In “Output - included information”
- “Set of columns to include in the variant report table”:
Custom (report user-specified columns)
- “Choose columns to include in the report”:
- param-check “alternative allele frequency (max_aaf_all)”
- “Additional columns (comma-separated)”:
chrom, start, ref, alt, impact, gene, clinvar_sig, clinvar_disease_name, clinvar_gene_phenotype, rs_idsComment: Variant- vs gene-centric ClinVar informationMost annotation data offered by GEMINI is variant-centric, i.e., annotations reported for a variant are specific to this exact variant. A few annotation sources, however, also provide gene-centric information, which applies to the gene affected by a variant, not necessarily the variant itself.
In the case of ClinVar, the annotation fields/columns
clinvar_sigandclinvar_disease_namerefer to the particular variant, butclinvar_gene_phenotypeprovides information about the affected gene.Including the gene phenotype in the report can be crucial because a gene may be well known to be disease-relevant, while a particular variant may not have been clinically observed or been reported before.
QuestionFrom the GEMINI reports you generated, can you identify the most likely candidate variant responsible for the child’s disease?
While only demonstrating command line use of GEMINI, the following tutorial slides may give you additional ideas for variant queries and filters:
Conclusion
It was not hard to find the most likely causative mutation for the child’s disease (you did find it, right?).
Even though it will not always provide as strong support for just one specific causative variant, analysis of whole-exome sequencing data of family trios (or other related samples) can often narrow down the search for the cause of a genetic disease to just a very small, manageable set of candidate variants, the relevance of which can then be addressed through standard methods.
You've Finished the Tutorial
Key points
Exome sequencing is an efficient way to identify disease-relevant genetic variants.
Freebayes is a good variant and genotype caller for the joint analysis of multiple samples. It is straightforward to use and requires only minimal processing of mapped reads.
Variant annotation and being able to exploit genotype information across family members is key to identifying candidate disease variants. SnpEff and GEMINI, in particular, are powerful tools offered by Galaxy for that purpose.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Wolfgang Maier, Bérénice Batut, Torsten Houwaart, Anika Erxleben, Björn Grüning, Exome sequencing data analysis for diagnosing a genetic disease (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/exome-seq/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{variant-analysis-exome-seq, author = "Wolfgang Maier and Bérénice Batut and Torsten Houwaart and Anika Erxleben and Björn Grüning", title = "Exome sequencing data analysis for diagnosing a genetic disease (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/exome-seq/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/variant-analysis/tutorials/exome-seq/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: bwa owner: devteam revisions: e188dc7a68e6 tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bwa owner: devteam revisions: 1dfc975b48d5 tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastqc owner: devteam revisions: 5ec9f6bceaee tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: freebayes owner: devteam revisions: 0d8581534009 tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_rmdup owner: devteam revisions: 586f9e1cdb2b tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bcftools_norm owner: iuc revisions: aaac3ed89508 tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bcftools_norm owner: iuc revisions: 5226c31ac6d3 tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: falco owner: iuc revisions: 959a14c1f2dd tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: gemini_inheritance owner: iuc revisions: 2c68e29c3527 tool_panel_section_label: Gemini tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: gemini_load owner: iuc revisions: 2270a8b83c12 tool_panel_section_label: Gemini tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: abfd8a6544d7 tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: 26ee5e11ecbe tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_view owner: iuc revisions: 5826298f6a73 tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_view owner: iuc revisions: 5ec526530725 tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: snpeff owner: iuc revisions: 6322be79bd8e tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: snpeff owner: iuc revisions: 3aae4f16ac9e tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: snpeff owner: iuc revisions: 3aae4f16ac9e tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: trimmomatic owner: pjbriggs revisions: b9aaed85cbd1 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/