Mapeo
| Autores/as |
|
| Traducción |
|
| Editores/as |
|
| Revisores/as |
|
Descripción GeneralPreguntas:
Objetivos:
¿Qué es el mapeo?
¿Qué dos cosas son cruciales para un mapeo correcto?
¿Qué es BAM?
Requisitos:
Correr una herramienta para mapear lectuas a un genoma de referencia
Explicar qué es un fichero BAM y qué contiene
Utilizar un buscador de genomas para entender tus datos
Duración estimada: 1 horaNivel: Introductorio IntroductoryMateriales de apoyo:
- Diapositivas
- Conjuntos de datos
- Flujos de trabajos
- FAQs
- video Recordings
- instances Disponible en estas instancias de Galaxy
Published: Mar 30, 2026Última modificación: Mar 30, 2026Licencia: El contenido de este tutorial tiene la licencia Creative Commons Attribution 4.0 International License. GTN Framework tiene licencia del MIT MITrating Rating: 3.0 (1 recent ratings, 1 all time)version Revision: 1
La secuenciación produce una colección de secuencias sin contexto genómico. No sabemos a qué parte del genoma corresponden las secuencias. El mapeo de las lecturas de un experimento con un genoma de referencia es un paso clave en el análisis moderno de datos genómicos. Con el mapeo, las lecturas se asignan a una ubicación específica en el genoma y se puede obtener información como el nivel de expresión de los genes.
Las lecturas no vienen con información de posición, por lo que no sabemos de qué parte del genoma provienen. Tenemos que utilizar la secuencia de la propia lectura para encontrar la región correspondiente en la secuencia de referencia. Pero la secuencia de referencia puede ser bastante larga (unos 3.000 millones de bases en el caso del ser humano), por lo que encontrar una región que coincida es una tarea de enormes proporciones. Como nuestras lecturas son cortas, puede haber varios lugares igualmente probables en la secuencia de referencia desde los que podrían haberse leído. Esto es especialmente cierto en el caso de las regiones repetitivas.
En principio, podríamos hacer un análisis BLAST para averiguar dónde encajan mejor las piezas secuenciadas en el genoma conocido. Tendríamos que hacerlo para cada uno de los millones de lecturas de nuestros datos de secuenciación. Sin embargo, alinear millones de secuencias cortas de este modo puede llevar un par de semanas. Y no nos importa la correspondencia exacta entre bases (alineación). Lo que nos interesa es “de dónde proceden estas lecturas”. Este enfoque se denomina mapeo.
A continuación, procesaremos un conjunto de datos con el mapeador Bowtie2 y visualizaremos los datos con el programa IGV.
AgendaEn este tutorial, nos ocuparemos de:
Preparar los datos
Práctica: Carga de datos
Crea un nuevo historial para este tutorial y dale un nombre apropiado
Haz click sobre el icono new-history en la parte superior del panel de historiales.
- Haz clic sobre Unnamed history (o el nombre que tenga el historial sobre el que estás trabajando) (Haz clic para cambiar el nombre del historial) en la parte superior de tu panel de historial
- Escribe el nombre nuevo
- Pulsa Enter
Importe
wt_H3K4me3_read1.fastq.gzywt_H3K4me3_read2.fastq.gzde Zenodo o de la biblioteca de datos (pregunte a su instructor)https://zenodo.org/record/1324070/files/wt_H3K4me3_read1.fastq.gz https://zenodo.org/record/1324070/files/wt_H3K4me3_read2.fastq.gz
- Copia los enlaces
Abre el manejador de carga de datos de Galaxy (galaxy-upload (Upload) en la parte superior derecha del panel de herramientas)
- Selecciona ‘Pegar/Traer datos’ Paste/Fetch Data
Copia los enlaces en el campo de textos
Presiona ‘Iniciar’ Start
Close Cierra la ventana.
- Galaxy utiliza los URLs como nombres de forma predeterminada , así que los tendrás que cambiar a algunos que sean más útiles o informativos.
Como alternativa a cargar los datos desde una URL o desde su ordenador, los archivos también pueden estar disponibles desde una biblioteca de datos compartidos:
- Entra en Libraries (panel izquierdo)
- Navega a la carpeta correcta indicada por su instructor. - En la mayoría de los tutoriales de Galaxias los datos serán proporcionados en una carpeta llamada GTN - Material –> Topic name -> Tutorial name.
- Seleccione los archivos deseados
- Haz clic en Add to History galaxy-dropdown cerca de la parte superior y selecciona as Datasets en el menú desplegable
- En la ventana emergente, elige
- “Seleccionar historial “: el historial al que desea importar los datos (o crear uno nuevo)
- Haga clic en Import
Por defecto, Galaxy toma el enlace como nombre, así que renómbralos.
Cambie el nombre de los archivos a
reads_1yreads_2
- Haga clic en el galaxy-pencil icono lápiz del conjunto de datos para editar sus atributos
- En el panel central, cambie el campo Name
- Haga clic en el botón Save

Acabamos de importar en Galaxy los archivos FASTQ correspondientes a los datos de fin de pares que pudimos obtener directamente de una instalación de secuenciación.
Durante la secuenciación se introducen errores, como la llamada de nucleótidos incorrectos. Los errores de secuenciación pueden sesgar el análisis y dar lugar a una interpretación errónea de los datos. El primer paso para cualquier tipo de datos de secuenciación es siempre comprobar su calidad.
Hay un tutorial dedicado al control de calidad de los datos de secuenciación. No vamos a repetir los pasos allí. Debería seguir el tutorial y aplicarlo a sus datos antes de continuar.
Mapear lecturas en un genoma de referencia
El mapeo de lecturas es el proceso para alinear las lecturas en un genoma de referencia. Un mapeador toma como entrada un genoma de referencia y un conjunto de lecturas. Su objetivo es alinear cada lectura en el conjunto de lecturas en el genoma de referencia, permitiendo desajustes, indels y el recorte de algunos fragmentos cortos en los dos extremos de las lecturas:
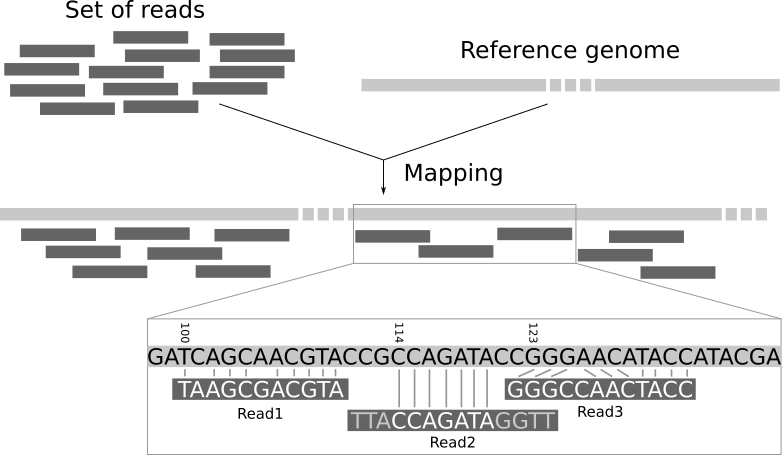 Open image in new tab
Open image in new tabNecesitamos un genoma de referencia para mapear las lecturas.
Preguntas
- ¿Qué es un genoma de referencia?
- Por cada organismo modelo, muchos genomas de referencia pueden estar disponibles (p.e.
hg19yhg38para humanos). ¿A qué corresponden?- ¿Qué genoma de referencia deberíamos usar?
- Un genoma de referencia (o ensamblado de referencia) es un conjunto de secuencias de ácidos nucleicos ensambladas como un ejemplo representativo del material genético de una especie. Dado que suelen ensamblarse a partir de la secuenciación de diferentes individuos, no representan con exactitud el conjunto de genes de un solo organismo, sino un mosaico de distintas secuencias de ácidos nucleicos provenientes de cada individuo.
- A medida que disminuye el costo de la secuenciación del ADN y surgen nuevas tecnologías de secuenciación de genomas completos, se generan cada vez más secuencias genómicas. Con estas nuevas secuencias, se construyen nuevas alineaciones y se mejoran los genomas de referencia (con menos huecos, corrección de errores en la secuencia, etc.). Los distintos genomas de referencia corresponden a las diferentes versiones publicadas, conocidas como “ensamblados” o builds.
- Estos datos provienen del ChIP-seq de ratones, por lo que utilizaremos mm10 (Mus musculus).
Actualmente, existen más de 60 mapeadores diferentes, y su número sigue creciendo. En este tutorial, utilizaremos Bowtie2, una herramienta de código abierto rápida y eficiente en memoria, particularmente buena para alinear lecturas de secuenciación de entre 50 y 1.000 bases con genomas relativamente largos.
Práctica: Mapeo con Bowtie2
- Bowtie2 ( Galaxy version 2.4.2+galaxy0) con los siguientes parámetros
- “Is this single or paired library “:
Paired-end
- param-file “FASTA/Q file #1”:
reads_1- param-file “FASTA/Q file #2”:
reads_2“Do you want to set paired-end options?”:
NoDebería echar un vistazo a los parámetros, especialmente la orientación de la pareja si la conoce. Pueden mejorar la calidad del mapeo de extremos pareados.
- “¿Will you select a reference genome from your history or use a built-in index?”:
Use a built-in genome index
- “Select reference genome “:
Mouse (Mus musculus): mm10“Select analysis mode “:
Default setting onlyDebería echar un vistazo a los parámetros no predeterminados e intentar comprenderlos. Pueden tener un impacto en el mapeo y mejorarlo.
- “Save the bowtie2 mapping statistics to the history “:
Yes- Inspeccione el archivo
mapping statshaciendo clic en el galaxy-eye (ojo)
Preguntas
- ¿Qué información se proporciona aquí?
- ¿Cuántas lecturas se han mapeado exactamente 1 vez?
- ¿Cuántas lecturas se han mapeado más de 1 vez? ¿Cómo es posible? ¿Qué debemos hacer con ellas?
- ¿Cuántos pares de lecturas no se han mapeado? ¿Cuáles son las causas?
- La información dada aquí es cuantitativa. Podemos ver cuántas secuencias están alineadas. No nos dice nada sobre la calidad.
- ~90% de las lecturas se han alineado exactamente 1 vez
- ~7% de las lecturas se han alineado concordantemente >1 veces. Estas se denominan lecturas multimapeadas. Puede ocurrir debido a repeticiones en el genoma de referencia (múltiples copias de un gen, por ejemplo), especialmente cuando las lecturas son pequeñas. Es difícil decidir de dónde proceden estas secuencias, por lo que la mayoría de los pipelines las ignoran. Compruebe siempre las estadísticas para asegurarse de no descartar demasiada información en los análisis posteriores.
- ~3% de pares de lecturas no se han mapeado porque
- ambas lecturas del par alineadas pero sus posiciones no concuerdan con el par de lecturas (
aligned discordantly 1 time)- las lecturas de estos pares están mapeadas (
aligned >1 timesenpairs aligned 0 times concordantly or discordantly)- se mapea una lectura de estos pares pero no la lectura emparejada (
aligned exactly 1 timeenpairs aligned 0 times concordantly or discordantly)- el resto no está mapeado en absoluto
La comprobación de las estadísticas de mapeo es un paso importante antes de continuar con cualquier análisis. Hay varias fuentes potenciales de errores en el mapeo, incluyendo (pero no limitado a):
- Artefactos de la reacción en cadena de la polimerasa (PCR): Muchos métodos de secuenciación de alto rendimiento (HTS) implican uno o varios pasos de PCR. Los errores de PCR se mostrarán como desajustes en la alineación, y especialmente los errores en las primeras rondas de PCR aparecerán en múltiples lecturas, sugiriendo falsamente una variación genética en la muestra. Un error relacionado son los duplicados de PCR, en los que el mismo par de lecturas aparece varias veces, lo que distorsiona los cálculos de cobertura en el alineamiento.
- Errores de secuenciación: La máquina de secuenciación puede hacer una llamada errónea, ya sea por razones físicas (por ejemplo, aceite en un portaobjetos Illumina) o debido a las propiedades del ADN secuenciado (por ejemplo, homopolímeros). Como los errores de secuenciación suelen ser aleatorios, pueden filtrarse como lecturas únicas durante la llamada de variantes.
- Errores de mapeo: El algoritmo de mapeo puede mapear una lectura a una localización incorrecta en la referencia. Esto ocurre a menudo alrededor de repeticiones u otras regiones de baja complejidad.
Por lo tanto, si las estadísticas de mapeo no son buenas, debe investigar la causa de estos errores antes de continuar con sus análisis.
Después de esto, debería echar un vistazo a las lecturas e inspeccionar el archivo BAM donde se almacenan los mapeos de lecturas.
Inspección de un archivo BAM
Un fichero BAM (Binary Alignment Map) es un archivo binario comprimido que almacena las secuencias leídas, indicando si han sido alineadas a una secuencia de referencia (por ejemplo, un cromosoma) y, en caso afirmativo, la posición en la secuencia de referencia donde han sido alineadas.
Práctica: Inspeccionar un BAM/SAM file
- Inspecciona el param-file output de Bowtie2 tool
Un fichero BAM (o SAM, la versión sin comprimir) consiste de:
- Una sección de cabecera (las líneas que comienzan con
@) que contiene metadatos, en particular los nombres y longitudes de los cromosomas (líneas que comienzan con el símbolo@SQ). -
Una sección de alineamiento que consiste en una tabla con 11 campos obligatorios, además de un número variable de campos opcionales:
Col Campo Tipo Breve descripción 1 QNAME String Query template NAME 2 FLAG Integer Bitwise FLAG 3 RNAME String References sequence NAME 4 POS Integer 1- based leftmost mapping POSition 5 MAPQ Integer MAPping Quality 6 CIGAR String CIGAR String 7 RNEXT String Ref. name of the mate/next read 8 PNEXT Integer Position of the mate/next read 9 TLEN Integer Observed Template LENgth 10 SEQ String Segment SEQuence 11 QUAL String ASCII of Phred-scaled base QUALity+33
Preguntas
- ¿Qué información se encuentra en un fichero BAM/SAM?
- ¿Cuál es la información adicional en comparación a un archivo FASTQ?
- Información de calidad y de secuencias, como un FASTQ
- Información de mapeo, localización de la lectura en el cromosoma, calidad del mapeo, etc.
El archivo BAM incluye mucha información sobre cada lectura, en particular sobre la calidad del mapeo.
Práctica: Resumen de la calidad del mapeo
- Samtools Stats ( Galaxy version 2.0.2+galaxy2) con los siguientes parámetros
- param-file “BAM file “:
aligned reads(salida de Bowtie2 tool)- “Use reference sequence “:
Locally cached/Use a built-in genome
- “Use genome”:
Mouse (Mus musculus): mm10 Full- Inspeccionar el archivo param-file archivo
Stats
Preguntas
- ¿Cuál es la proporción de desajustes en las lecturas mapeadas cuando se alinean con el genoma de referencia?
- ¿Qué representa la tasa de error?
- ¿Cuál es la calidad media? ¿Cómo se representa?
- ¿Cuál es el tamaño medio de las inserciones?
- ¿Cuántas lecturas tienen una puntuación de calidad de mapeo inferior a 20?
- Hay ~21.900 discordancias para ~4.753.900 bases mapeadas, lo que en promedio produce ~0,005 discordancias por bases mapeadas.
- La tasa de error es la proporción de emparejamientos erróneos por bases mapeadas, por lo que la proporción calculada justo antes.
- La calidad media es la puntuación de calidad media del mapeo. Es una puntuación Phred como la utilizada en el archivo FASTQ para cada nucleótido. Pero aquí la puntuación no es por nucleótido, sino por lectura y representa la probabilidad de la calidad del mapeo.
- El tamaño de la inserción es la distancia entre las dos lecturas de los pares.
- Para obtener la información:
- Filter BAM ( Galaxy version 2.5.2+galaxy2) con un filtro para mantener sólo las lecturas con una calidad de mapeo >= 20
- Samtools Stats ( Galaxy version 2.0.5) en la salida de Filter
Antes del filtrado: 95.412 lecturas y después del filtrado: 89.664 lecturas.
Visualización mediante un navegador del genoma
IGV
El Visor de Genómica Integrativa (IGV) es una herramienta de visualización de alto rendimiento para la exploración interactiva de grandes conjuntos de datos genómicos integrados. Es compatible con una amplia variedad de tipos de datos, incluidos los datos de secuencias basadas en arrays y de próxima generación, y las anotaciones genómicas. A continuación, la utilizaremos para visualizar las lecturas mapeadas.
Práctica: Visualización de lecturas en IGVHay dos maneras de correrlo:
- Si tienes IGV instalado (o quieres instalarlo):
- Instala IGV
- Inicia IGV en tu ordenador
- Expande el param-file output de Bowtie2 tool
- Haz click en
localendisplay with IGVpara cargar tus lecturas en el navegador de IGV- Si no tienes IGV
- Click en el
Mouse mm10(o en el organismo correcto) endisplay with IGVpara cargar tus lecturas en el navegador de IGV- Zoom en
chr2:98,666,236-98,667,473
Las lecturas tienen una dirección: se mapean a la hebra directa o a la hebra inversa, respectivamente. Al pasar el cursor sobre una lectura, se muestra información adicional.
Preguntas
- ¿Qué podría significar si una barra en la vista de cobertura está colorada?
- ¿Cuál podría ser la razón para que una lectura sea blanca en vez de gris?
- Si un nucleótido difiere de la secuencia de referencia en más del 20 % de las lecturas ponderadas por calidad, IGV colorea la barra en proporción al número de lecturas de cada base.
- Tienen una calidad de mapeo igual a cero. La interpretación de esta calidad de mapeo depende del alineador utilizado, ya que algunos alineadores comunes usan esta convención para marcar lecturas con alineamientos múltiples. En ese caso, la lectura también se alinea en otra ubicación con una calidad de alineamiento igualmente buena. También es posible que la lectura no pueda ubicarse de forma única, aunque las otras ubicaciones alternativas no necesariamente tengan una calidad de alineamiento igual de buena.
Comentario: Trucos para IGV
- Debido a que el número de lecturas en una región puede ser muy elevado, el visor IGV muestra por defecto solo las lecturas que caen dentro de una pequeña ventana. Este comportamiento puede modificarse en IGV desde
view > Preferences > Alignments.Si el genoma de interés no está ahí comprueba si está disponible en More…. Si este no es el caso, puedes añadirlo manualmente a través del menú Genomes -> Load Genome from…
Una descripción general de la interfaz de IGV está disponible aquí: IGV Browser description

JBrowse
JBrowse ( Galaxy version 1.16.11+galaxy0) es un navegador del genoma alternativo, basado en la web. Mientras que IGV es un software que debe descargarse y ejecutarse, las instancias de JBrowse son sitios web alojados en línea que proporcionan una interfaz para navegar por los datos genómicos. Lo utilizaremos para visualizar las lecturas mapeadas.
Práctica: Visualización de lecturas JBrowse
- JBrowse ( Galaxy version 1.16.11+galaxy1) con los siguientes parámetros:
- “Reference genome to display”: Use a built-in genome
- “Select a reference genome”:
mm10- “JBrowse-in-Galaxy Action”:
New JBrowse Instance- “Insert Track Group”:
- “Insert Annotation Track”:
- “Track Type”: BAM Pileups
- “BAM Track Data”:
aligned reads(output de Bowtie2 tool)- “Autogenerate SNP Track”: Yes
- “Track Visibility”: On for new users
- Visualiza the dataset galaxy-eye
- Zoom en el
chr2:98,666,236-98,667,473
Comentario: SlowEsto puede tardar uno o dos minutos en ejecutarse, dependiendo de los recursos de tu instancia de entrenamiento. Toma tiempo porque el servidor construye un pequeño sitio web para ti y preprocesa el genoma de referencia en un formato más eficiente. Si quisieras compartir esto con tus colegas, podrías descargar este conjunto de datos y colocarlo directamente en tu propio servidor web.
Las lecturas tienen una dirección: se alinean a la hebra directa o a la hebra inversa, respectivamente. Al hacer clic sobre una lectura, se muestra información adicional.
Preguntas
- ¿Qué significan la gota y la línea en el SNP autogenerado?
- ¿Qué significan las lecturas de diferentes colores?
- Si suficientes lecturas tienen un valor diferente, se marca con un ícono en forma de lágrima. La gráfica de cobertura se representa en altura según el porcentaje de lecturas con una llamada diferente en esa posición.
Códigos de colores:
Colour Meaning Original red Forward strand Original blue Reverse strand Hard red Forward strand, missing mate Hard Blue Reverse strand, missing mate Light red Forward strand not proper Light blue Reverse strand, not proper Black Forward, diff chr Grey Reverse, diff chr Grey No strand
Conclusión
Tras el control de calidad, el mapeo es un paso importante en la mayoría de los análisis de datos de secuenciación (RNA-Seq, ChIP-Seq, etc.) para determinar en qué parte del genoma se originaron nuestras lecturas y utilizar esta información para los análisis posteriores.
You've finished the tutorial
Puntos clave
¡Conoce tus datos!
El mapeo no es trivial.
Existen muchos algoritmos de mapeo; depende de tus datos cuál elegir.
Preguntas frecuentes
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Retroalimentación
¿Utilizaste este material como instructor? Cuéntanos tu experiencia.
¿Has usado este material como aprendiz o estudiante? Haz click en el formulario a continuación para dejarnos tu opinión

Cómo citar este tutorial
- Joachim Wolff, Bérénice Batut, Helena Rasche, Mapeo (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/mapping/tutorial_ES.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{sequence-analysis-mapping, author = "Joachim Wolff and Bérénice Batut and Helena Rasche", title = "Mapeo (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/mapping/tutorial_ES.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Referencias
These individuals or organisations provided funding support for the development of this resource

¿Quieres expandir tus conocimientos?Continúa tu aprendizaje completando uno de nuestros entrenamientos recomendados:
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/sequence-analysis/tutorials/mapping/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: trim_galore owner: bgruening revisions: 949f01671246 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bamtools_filter owner: devteam revisions: cb20f99fd45b tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bamtools_filter owner: devteam revisions: 993b19f20c76 tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bowtie2 owner: devteam revisions: f76cbb84d67f tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastqc owner: devteam revisions: e7b2202befea tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_stats owner: devteam revisions: 24c5d43cb545 tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_stats owner: devteam revisions: 541082d03bef tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_stats owner: devteam revisions: 57934e90e27c tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: jbrowse owner: iuc revisions: 4542035c1075 tool_panel_section_label: Graph/Display Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: jbrowse owner: iuc revisions: a6e57ff585c0 tool_panel_section_label: Graph/Display Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: c1a4b5f3b432 tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/