Mapping
Contributors
Questions
What is mapping (alignment)?
What is the BAM format?
How can we view aligned sequences?
Objectives
Understand the basic concept of mapping
Learn about factors influencing alignment
See a genome browser used to better understand your aligned data
Example NGS pipeline
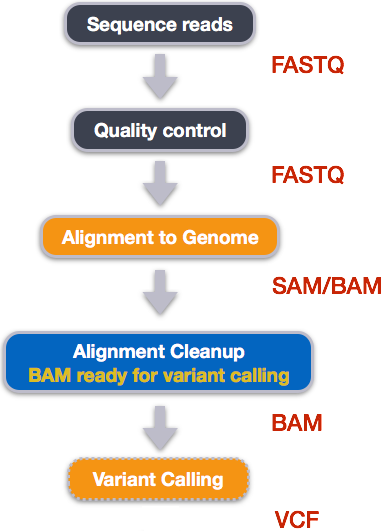
A high level view of a typical NGS bioinformatics workflow
Speaker Notes
- Mapping step occurs if a reference genome is available for the organism of interest
- else: de-novo assembly
- Variant calling step is just an example, after mapping can do many steps
- Structural Variants / Fusion genes
- Differential Gene expression
- Alternative Splicing
- ..
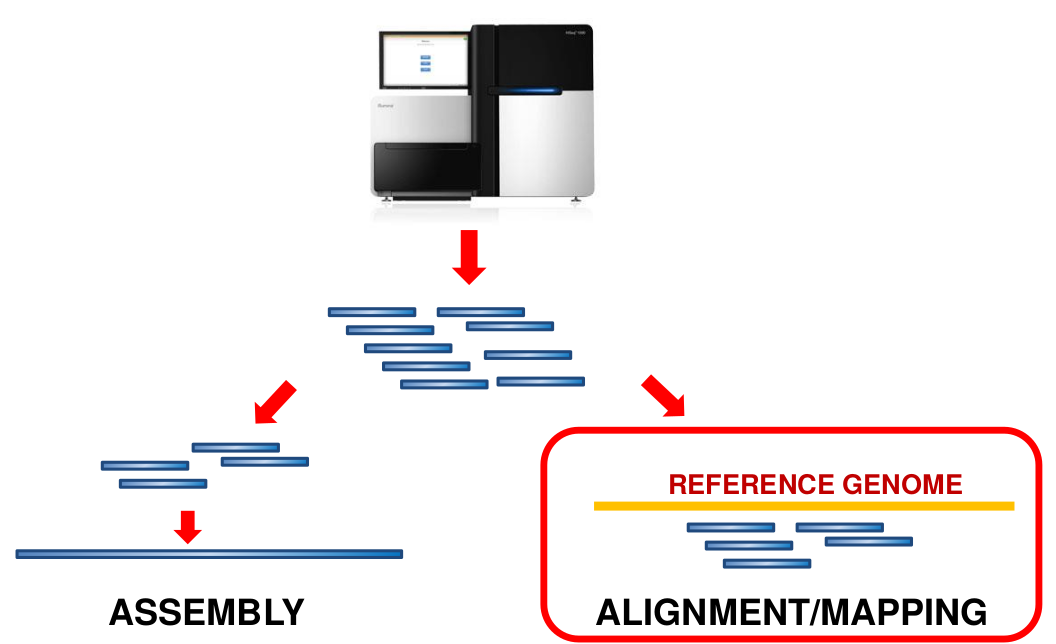
What is mapping?
.pull-left[
 ]
]
.pull-right[
-
Short reads must be combined into longer fragments
-
Mapping: use a reference genome as a guide
-
De-novo assembly: without reference genome ]
Speaker Notes
- Mapping is also referred to as alignment
- Short reads produced by sequencer must be combined into larger contigs
- e.g. reconstruct the chromosomes
- mapping uses a reference genome as a guide
- can subsequently find where our sample differs from reference (variants)
- This tutorial only deals with mapping/alignment
- There are other tutorials available for de-novo assembly
class: top
Sequence alignment
- Determine position of short read on the reference genome
Reference: . . . A A C G C C T T . . . Read: A G G G G C C T T
Speaker Notes
- Consider situation where we must map this (short) read to this (long) reference
- e.g. human genome ~ 3.2 billion base pairs
- We scan the reference genome until we find an area that’s similar to our read
- This area looks pretty similar, but not quite identical..
class: top
Sequence alignment
- Determine position of short read on the reference genome
Reference: . . . A A - C G C C T T . . . | = match . | : - : | | | | | : = mismatch Read: A G G G G C C T T - = gap
Speaker Notes But if we introduce gaps and allow for some mismatches in bases, this matches up pretty well..
–
-
Read could align to multiple places .center[.image-50[
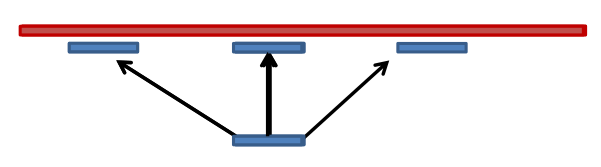 ]]
]] - How to handle multi-mapped reads? Depends on tool:
- Map to best region (but what is “best”? And what about ties?)
- Map to all regions
- Map to one region randomly
- Discard read
- How do we determine best region?
- Assign alignment score to every mapping
Speaker Notes
Some reads may map to multiple locations
- repeat regions, short reads, highly variable regions, sequencing errors, ..
We want a way to determine best alignment if none are perfect matches..
class: top
Alignment Scoring (basics)
- Reward for a match (e.g. +10), penalty for a mismatch (e.g. -5)
- Penalty for gaps
- Linear: every gap same penalty (e.g. -5)
- Affine: gap open vs gap extend (e.g. -5 and -1)
- Different tools use different scoring values (and give different results)
.center[
.image-25[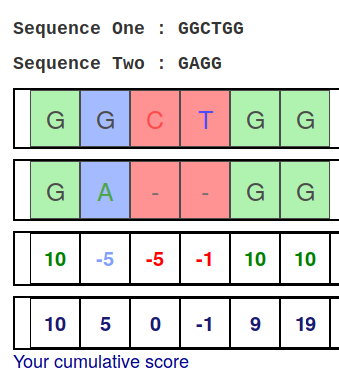 ]
]
Example (with affine gap penalty) ]
Speaker Notes
- Each locus get scored independently (first row of scores in example)
- Scores from all loci are added up (cumulative score row)
-
Final score for entire alignment in this example is 19
- These reward and penalty values are just examples and will vary
class: top
Alignment Scoring (advanced)
- Base quality
- Mismatch of low-confidence base: lower penalty
- Mismatch of high-confidence base: higher penalty
- Transitions vs transversions
- Transitions about 2x as frequent as transversions
.center[
.image-50[ 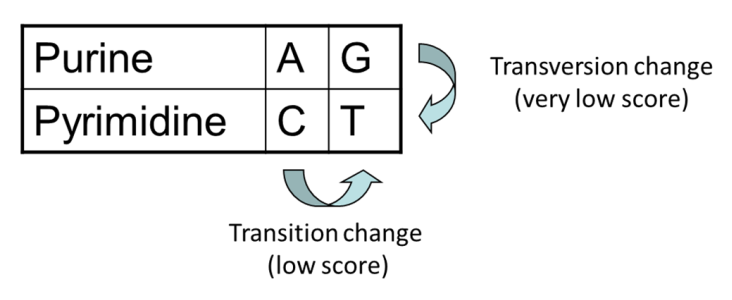 ]
.image-25[
]
.image-25[ 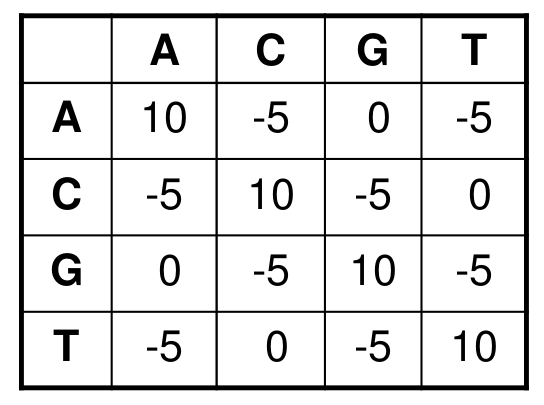 ]
]
]
]
- Knowledge about sequencing platform and biases
- Optimize for read length, error rate, homopolymer accuracy, etc..
.footnote[More information about mapping algorithms: 10.1089/cmb.2012.0022]
Speaker Notes Many more complexities may be considered, different tools make different choices
Transitions are more likely to occur in real sequences, so may give lower penalty than transversions
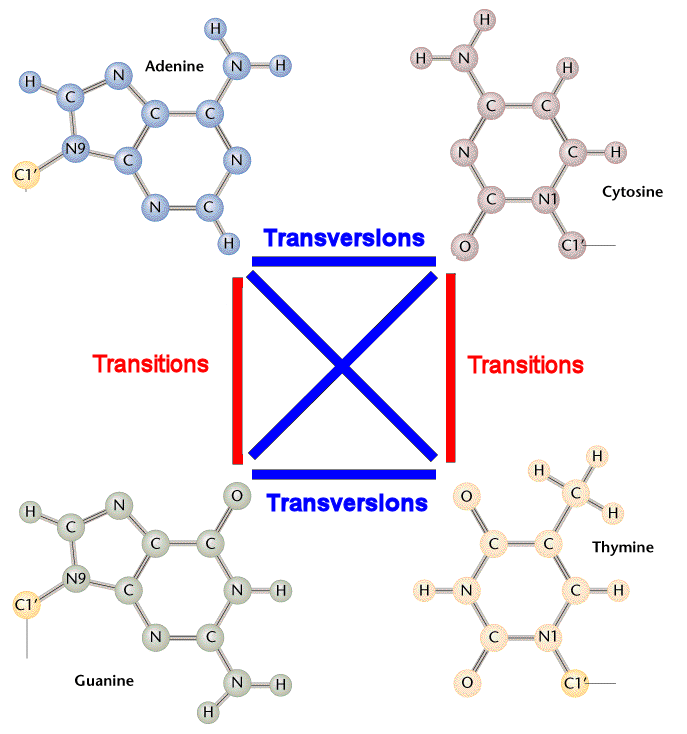
Transitions are interchanges of two-ring purines (A G) or of one-ring pyrimidines (C T): they therefore involve bases of similar shape.
Transversions are interchanges of purine for pyrimidine bases, which therefore involve exchange of one-ring and two-ring structures.
Looks easy but..
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
Speaker Notes Suppose we want to map this read (bottom) to this reference sequence (top)
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
Maybe like this? |
Speaker Notes This is one possibility, is it the only one?
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
Maybe like this? |
AAACAGTGAGAA |||-::|::||| AAA-TCTCTGAA |
Or this? |
Speaker Notes This is also a possible alignment. Not easy to say which is better.
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
Maybe like this? |
AAACAGTGAGAA |||-::|::||| AAA-TCTCTGAA |
Or this? |
AAACAGTGAGAA |||:-:|::||| AAAT-CTCTGAA |
Or..? |
Speaker Notes And a third option
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
Maybe like this? |
AAACAGTGAGAA |||-::|::||| AAA-TCTCTGAA |
Or this? |
AAACAGTGAGAA |||:-:|::||| AAAT-CTCTGAA |
Or..? |
AAACAGTCA-----GAA |||-----------||| AAA------TCTCTGAA |
What about this? |
Speaker Notes There is no one right way to do alignment
- Hard to say which of these is “better” or “worse”
- Just different choices, but all valid
Mapping is a non-trivial problem!
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | Tool |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
Novoalign |
AAACAGTGAGAA |||-::|::||| AAA-TCTCTGAA |
Ssaha2 |
AAACAGTGAGAA |||:-:|::||| AAAT-CTCTGAA |
BWA |
AAACAGTCA-----GAA |||-----------||| AAA------TCTCTGAA |
Complete Genomics |
Speaker Notes We didn’t just make these up, these real aligners gave these different results
class: top
Sequence Alignment
Reference: AAA CAGTGA GAA
Observed: AAA TCTCT GAA
| Alignment | Variant calls |
|---|---|
AAA-CAGTGAGAA |||-|--|::||| AAATC--TCTGAA |
ins T del AG sub GA -> CT |
AAACAGTGAGAA |||-::|::||| AAA-TCTCTGAA |
del C sub AG -> TC sub GA -> CT |
AAACAGTGAGAA |||:-:|::||| AAAT-CTCTGAA |
snp C -> T del A snp G -> C sub GA -> CT |
AAACAGTGA-----GAA |||-----------||| AAA------TCTCTGAA |
del CAGTGA ins TCTCT |
Speaker Notes Important: Mapping can affect downstream analysis!
These different mappings led to different variants, and hard to tell they are equivalent.
Try it yourself!
-
Lego time! Who wants to volunteer?
-
Or try this online sequence alignment game:
.image-75[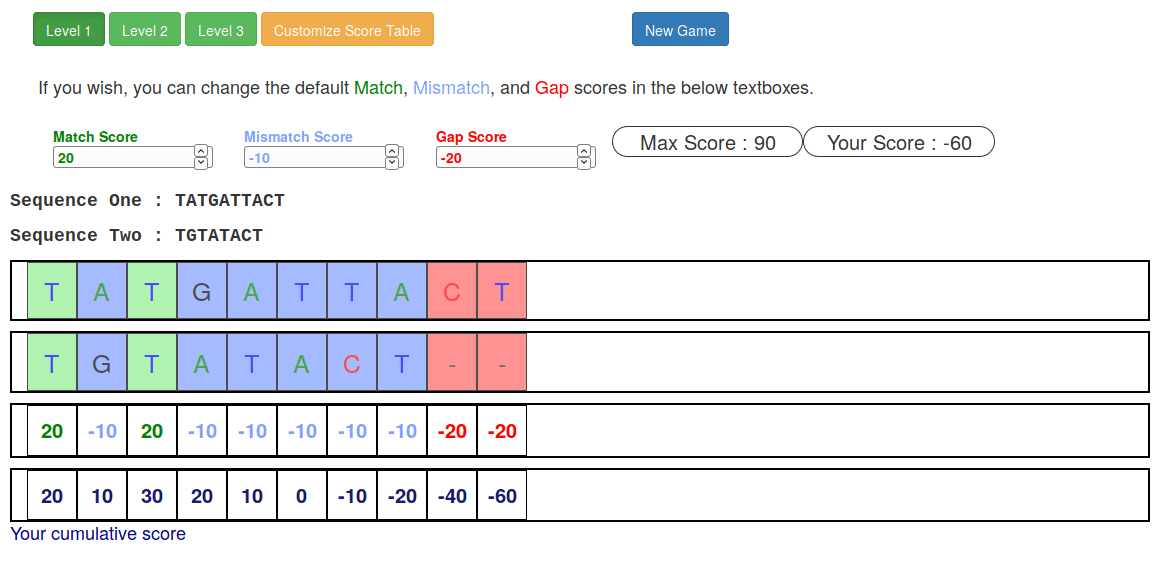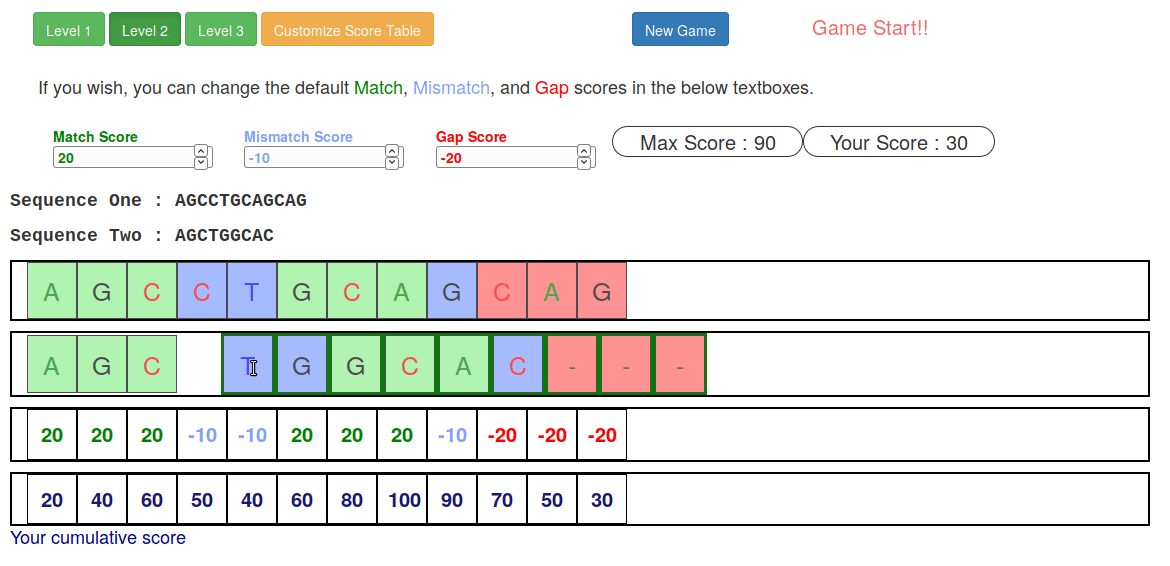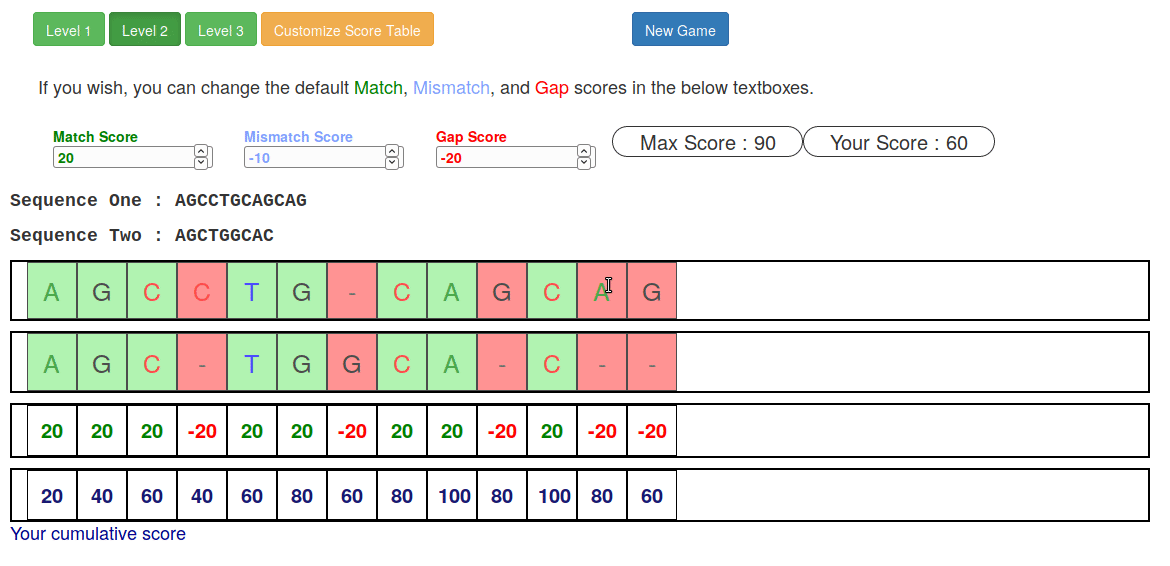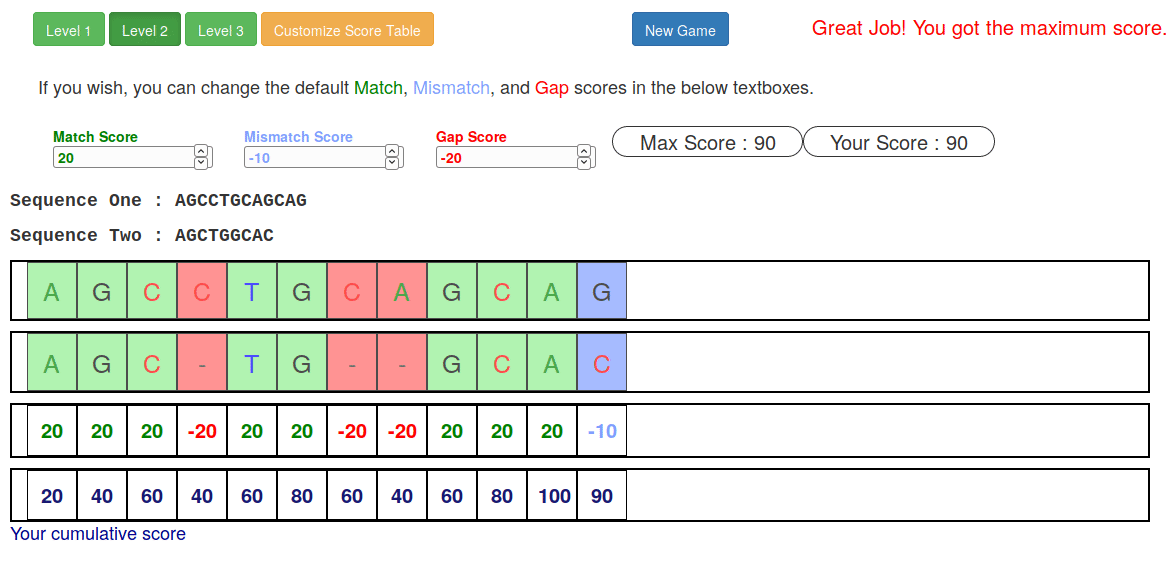 ]
]
.footnote[https://tinyurl.com/sequence-alignment]
Speaker Notes Can have learners play around with this alignment game now
Or use Lego bricks, each nucleotide a different colour
Paired-end sequencing
-
Sequencing: Cut longer fragments of DNA, sequence only the ends
.center[.image-90[
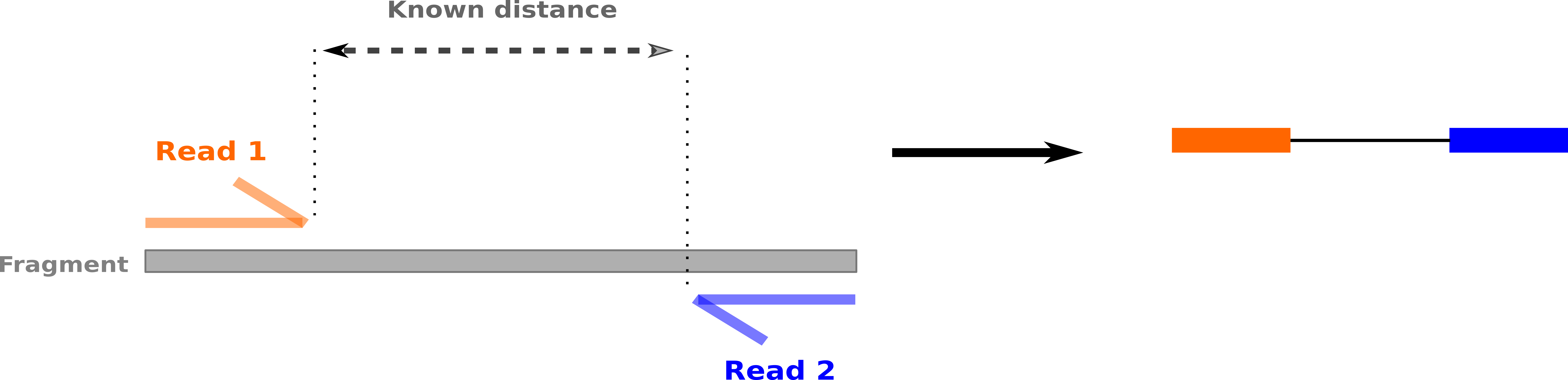 ]]
]] -
Mapping: known distance between reads improves accuracy
.center[.image-75[
 ]]
]]
Speaker Notes
- The fragments are too long to sequence entirely, but we can sequence the ends.
- Then we have the added information of how far apart these two reads must map
-
This improves our mapping
- For example for multi-mapped reads, or repeats (next slide)
class: top
Repeats
-
Multi-mapped reads (e.g. because of repeats) may now be resolved
-
Single-end:

Speaker Notes In the case of repeats, a single-end read alone would not have be enough for unique mapping..
–
-
Paired-end:
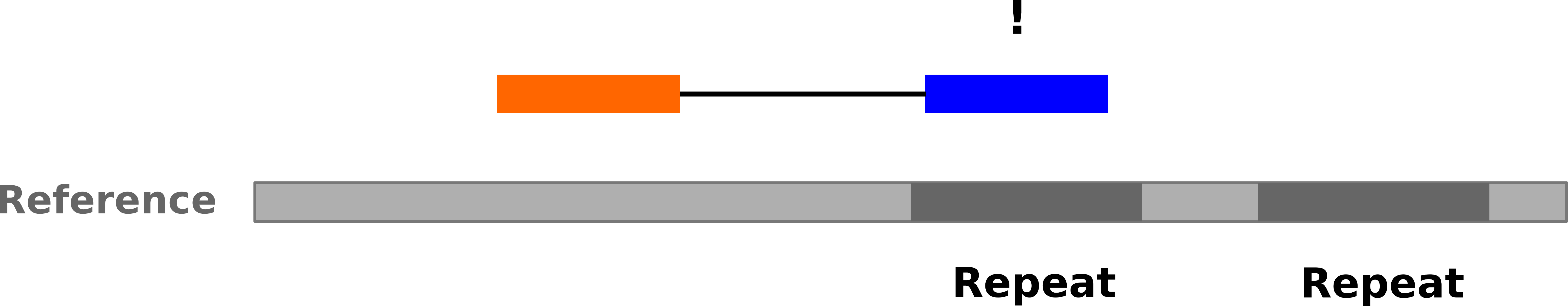
Speaker Notes But with the additional information provided by paired-end protocol (distance to mate), this can now be resolved..
class: top
InDels (Insertions / Deletions)
-
Discordant insert size may indicate insertion or deletion between reads
-
Deletions: Longer mapping distance than expected
.image-75[
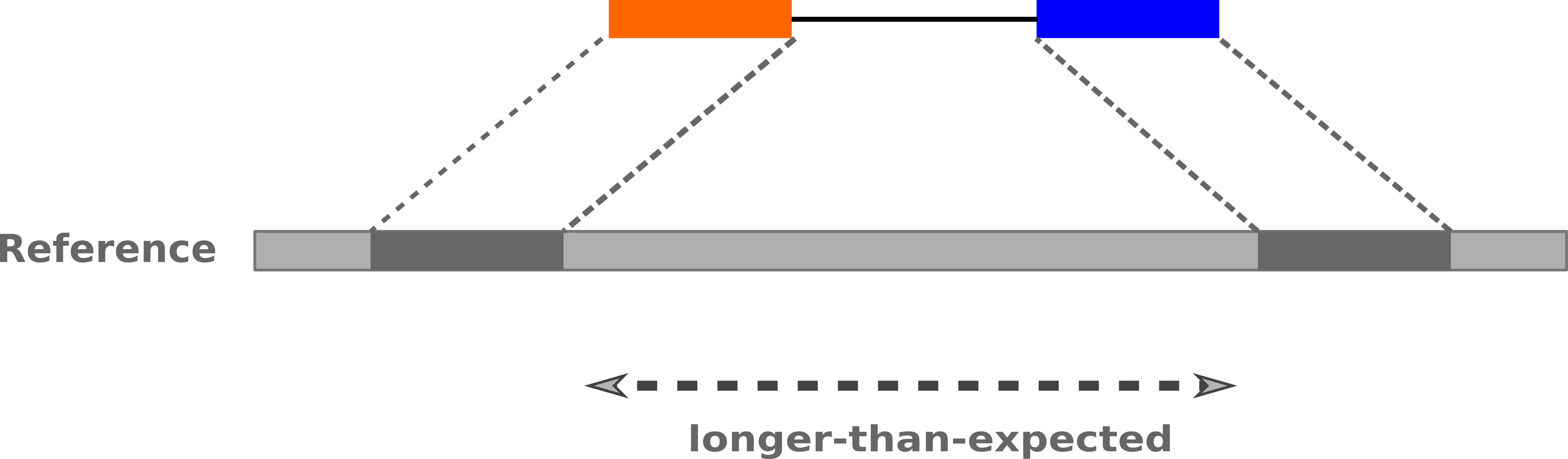 ]
]
–
-
Insertions: Shorter mapping distance than expected
.image-75[
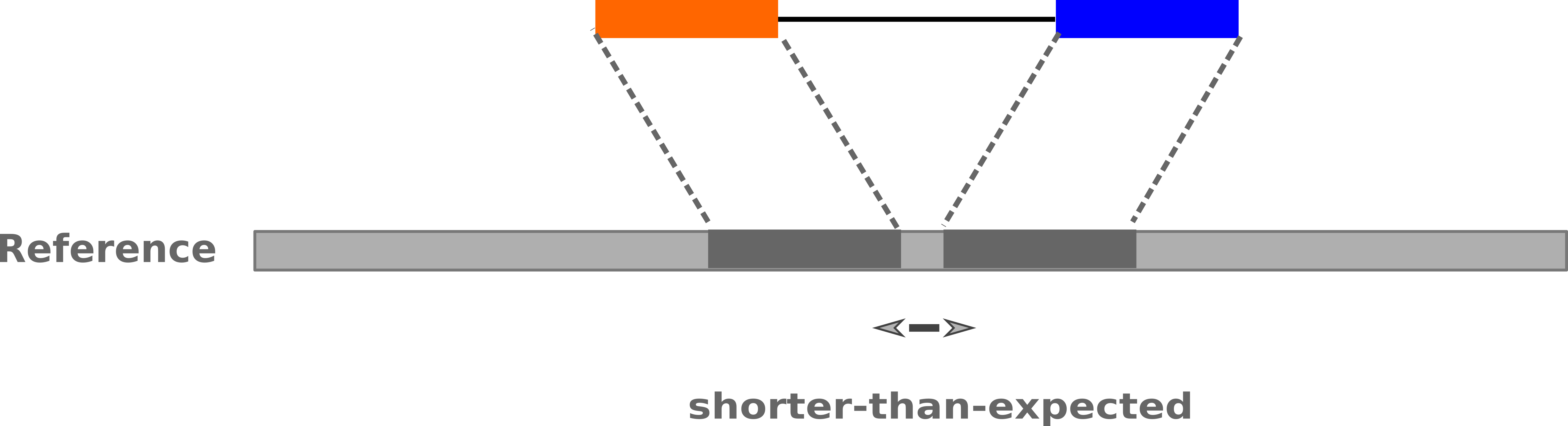 ]
]
Speaker Notes
-
Unexpected mapping distance between two reads in a pair may indicate a variant.
-
Exact location of variant unknown unless more reads covering the area
- Only know it it somewhere between the two reads
FAQ: “What about mate-pair sequencing?”
- Same concept as paired-end
- Much longer distance between ends
- Very different library prep
- Useful for detection of larger Structural Variations (SVs) / Fusion Genes
- longer than expected distance between mates: deletion in sample
- shorter than expected distance beetween mates: insertion in sample
- unexpected orientation of one mate: inversion in sample
class: top
Paired-end FASTQ files
- Sequencer produces two FASTQ files:
- Forward reads (usually
_1or_R1in file name) - Reverse reads (usually
_2or_R2in file name)
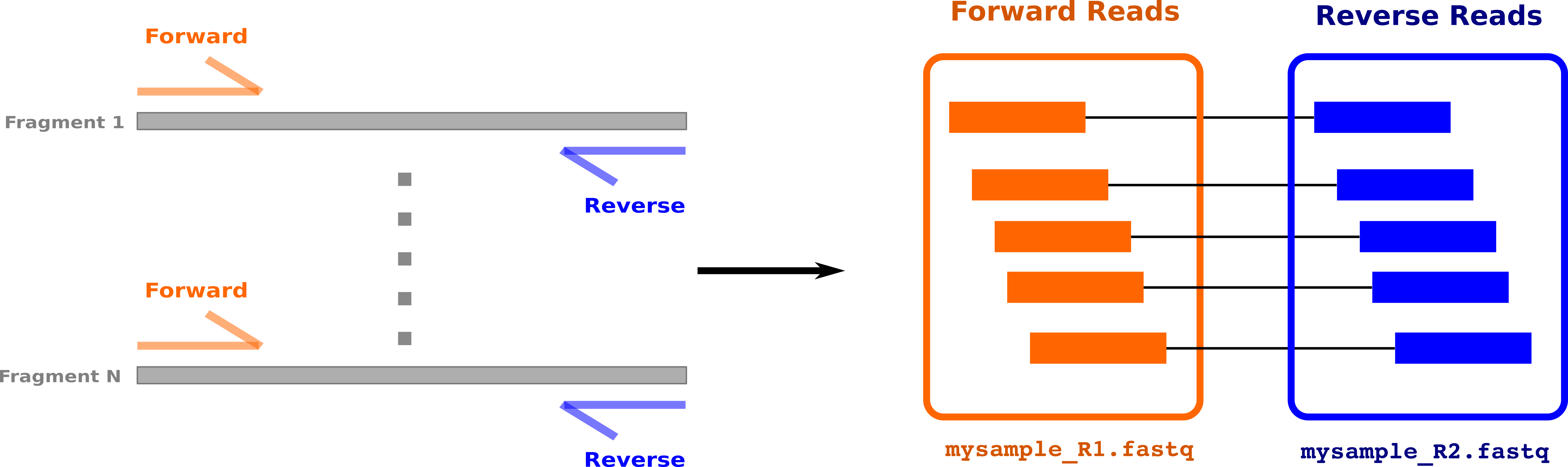
- Forward reads (usually
Speaker Notes When you have paired-end data, you will usually get 2 files.
- File names identical except for e.g.
_1/_2or_R1/_R2 - First file contains all the forward reads (“left” sides of pairs)
- Other file contains all the reverse reads
Pairing also visible in read names
/1/2at end or1:and2:in read ID
–
- Sometimes: One interleaved (or interlaced) FASTQ file
- Most tools require 2 separate files
- tool De-interlace tools in Galaxy for conversion
Speaker Notes Sometimes data can be in a single interleaved file (aka interlaced)
- alternating forward and reverse read
- de-interlace tools in Galaxy to convert this to two separate files
- because many tools require two separate files
class: top
Paired-end FASTQ files
- Order of reads matters!
Nth read in forward fileNth read in reverse file- Much faster than determining pairing by read names alone
- Always trim and filter together!
Speaker Notes Most tools blindly assume that first read in forward file is paired with first read in reverse file etc
Otherwise too slow
- for every read, worst case have to scan all reads in other file
- for files with millions of reads, that is millions ^ millions
When trimming and filtering, if a read is removed from one file, its mate must be removed from other one too!
Always trim together in paired-end mode!
–
.pull-left[ .red[
@PAIR-1 forward
GGGTGATGGCCGCTGCCGATGGCGTCAAAT
+
))%255CCF>>>>>>CCCCCCC65`IIII%
] .orange[
@PAIR-2 forward
GATTTGGGGTTCAAAGCAGTATCGATCAA
+
!''3((((^^d+))%%%++)(%%%%).1)
] .blue[
@PAIR-3 forward
TCGCACTCAACGCCCTGCATATGACAAGAC
+
A64;##=#B9=AAAAAAAAAA9#:AB95%^
]
mysample_R1.fastq
]
.pull-right[ .red[
@PAIR-1 reverse
AAGTTACCCTTAACAACTTAAGGGTTTTCA
+
fffddf`feedB`IABa)^%YBBBRTT\^d
] .orange[
@PAIR-2 reverse
AGCAGAAGTCGATGATAATACGCGTCGTTT
+
IIIIIII^^IIId`?III%IIIGII>IIII
] .blue[
@PAIR-3 reverse
AATCCATTTGTTCAACTCACAGTTTACCGT
+
9C;=;=<9@4868>9:67AA<9>65<=>59
]
mysample_R2.fastq
]
Speaker Notes
- Nth read in forward file belongs in a pair with Nth read in reverse file
- So red reads in this slide form a pair, orange ones, etc
class: top
Paired-end FASTQ files
- Order of reads matters!
Nth read in forward fileNth read in reverse file- Much faster than determining pairing by read names alone
- Always trim and filter together!
.pull-left[ .red[
@PAIR-1 forward
GGGTGATGGCCGCTGCCGATGGCGTCAAAT
+
))%255CCF>>>>>>CCCCCCC65`IIII%
] .left[] .orange[
@PAIR-2 forward
GATTTGGGGTTCAAAGCAGTATCGATCAA
+
!''3((((^^d+))%%%++)(%%%%).1)
] .blue[
@PAIR-3 forward
TCGCACTCAACGCCCTGCATATGACAAGAC
+
A64;##=#B9=AAAAAAAAAA9#:AB95%^
]
mysample_R1.fastq
]
.pull-right[
.red[
@PAIR-1 reverse
AAGTTACCCTTAACAACTTAAGGGTTTTCA
+
fffddf`feedB`IABa)^%YBBBRTT\^d
] .orange[
@PAIR-2 reverse
AGCAGAAGTCGATGATAATACGCGTCGTTT
+
IIIIIII^^IIId`?III%IIIGII>IIII
] .blue[
@PAIR-3 reverse
AATCCATTTGTTCAACTCACAGTTTACCGT
+
9C;=;=<9@4868>9:67AA<9>65<=>59
]
mysample_R2.fastq
]
Speaker Notes
- Important to always provide both files to trimming and filtering tools together
-
If a read in one file gets removed (e.g. because it is below quality threshold), but it’s mate is not, the pairing between the two files is no longer correct.
- If one half of pair is trimmed, the other
- also removed, or
-
put into separate “singletons” FASTQ file that some mappers can use
- FAQ:” why not look at read names to determine pairing?”
-
analysis would be much slower if for every read must scan (max) entire other file for mate, since often millions or reads (for whole-genome sequencing).
class: top
-
Paired-end FASTQ files
- Order of reads matters!
Nth read in forward fileNth read in reverse file- Much faster than determining pairing by read names alone
- Always trim and filter together!
.pull-left[ .red[
@PAIR-1 forward
GGGTGATGGCCGCTGCCGATGGCGTCAAAT
+
))%255CCF>>>>>>CCCCCCC65`IIII%
] .blue[
@PAIR-3 forward
TCGCACTCAACGCCCTGCATATGACAAGAC
+
A64;##=#B9=AAAAAAAAAA9#:AB95%^
] .green[
@PAIR-4 forward
AAACTTCGTAGGTCCATTTGACAGCGTGCA
+
6664%!!III^(=%3333^^d^d:#32333
]
mysample_R1.fastq
]
.pull-right[
.red[
@PAIR-1 reverse
AAGTTACCCTTAACAACTTAAGGGTTTTCA
+
fffddf`feedB`IABa)^%YBBBRTT\^d
] .orange[
@PAIR-2 reverse
AGCAGAAGTCGATGATAATACGCGTCGTTT
+
IIIIIII^^IIId`?III%IIIGII>IIII
] .blue[
@PAIR-3 reverse
AATCCATTTGTTCAACTCACAGTTTACCGT
+
9C;=;=<9@4868>9:67AA<9>65<=>59
]
mysample_R2.fastq
]
Speaker Notes By cutting the yellow read only from the forward reads file, but leaving the other side of pair in the other file, an incorrect pairing is now assumed by downstream tools
Choosing an Aligner
- Each tool makes different choices during alignment
- Choice of aligner may affect downstream results
- Default options may not be best for your data!
- Best tool for your data depends on many factors
- Type of experiment (e.g. DNA, RNA, Bisulphite)
- Sequencing platform
- Compute resources vs sensitivity
- Read characteristics (paired/single end, read length)
.center[
.image-40[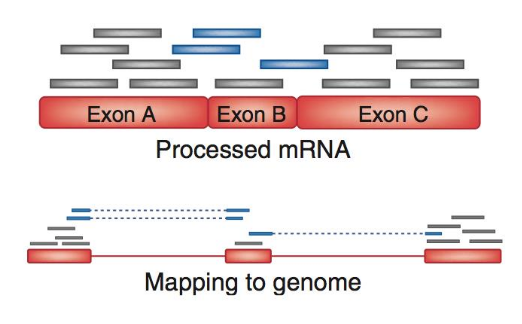 ]
]
.footnote[Figure: mapping of RNA-seq reads is different than DNA-seq]
]
]
.footnote[Figure: mapping of RNA-seq reads is different than DNA-seq]
Speaker Notes Choice of mapper depends on your experiment
- Some mappers are good for DNA but not RNA
- Some mappers do well in highly rearranged genomes (e.g. cancer), others less so
- Some mappers do well on some platforms but worse on others
- e.g. Oxford Nanopore with its long reads but high error rates
Or other factors
- STAR needs a LOT of RAM
- Do you need results fast? or accurate? (e.g. medical setting)
FAQ: “Why not map RNA reads to the transcriptome?”
- you can, and it is done, but then cannot find novel genes or alternative splicing
FAQ: “Why not BLAST or BLAT?”
- optimized for longer sequences
- not base quality aware
- too slow
Know your data!
“… there is no tool that outperforms all of the others in all the tests. Therefore, the end user should clearly specify [their] needs in order to choose the tool that provides the best results.” - Hatem et al BMC Bioinformatics 2013, 14:184
.footnote[ DOI: 10.1186/1471-2105-14-184 ]
Speaker Notes
Know the data you are working with and pick the right mapper and parameters for the job!
Not an easy task..
class: top
Mapping tools
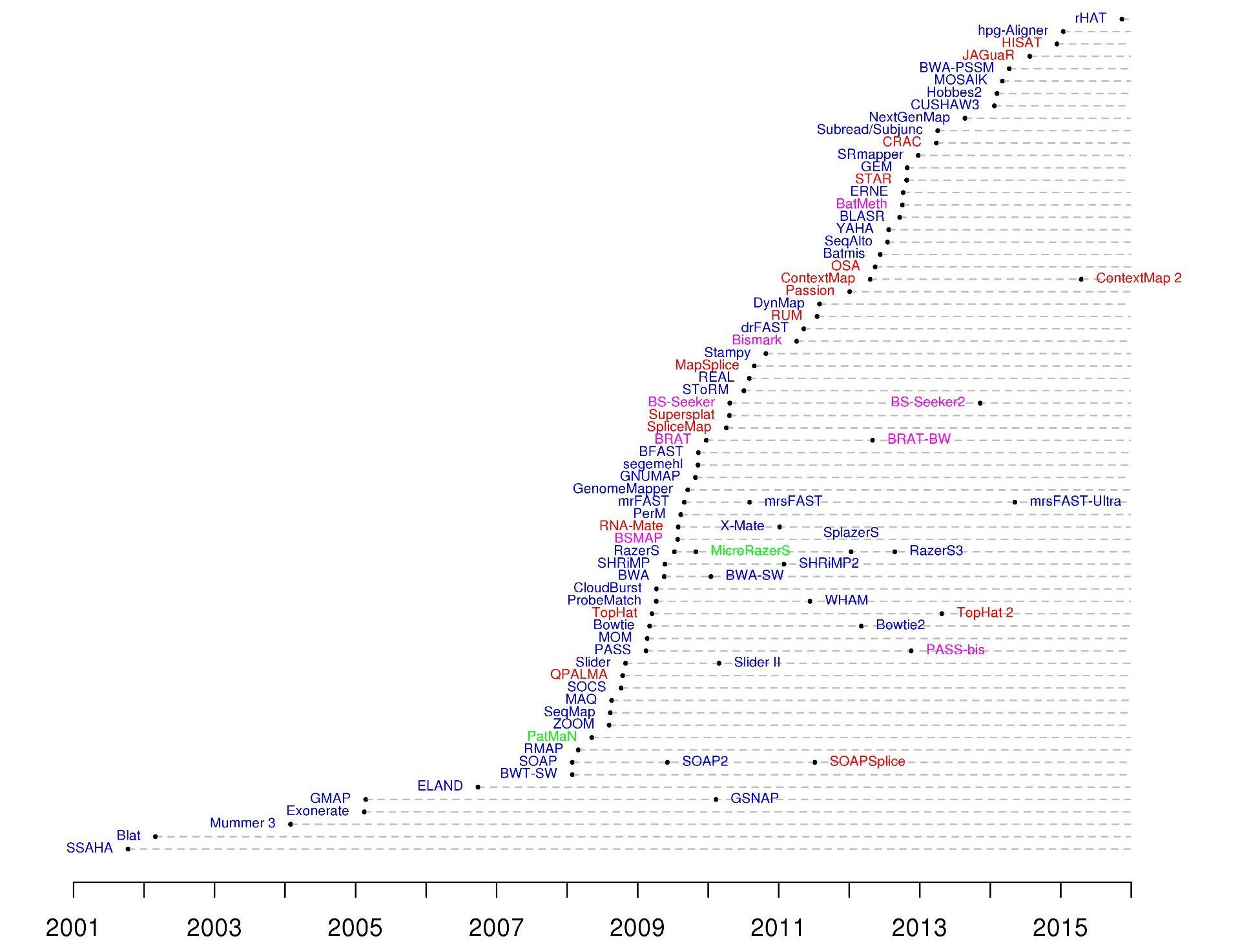
.footnote[60+ different mappers, many comparison papers. Figure from 10.1093/bioinformatics/bts605 ]
Speaker Notes
Many different tools available
Different strengths and weaknesses, comparison table in link
class: top
Mapping tools
| Mapping tool | Uses | Characteristics |
|---|---|---|
| HISAT2 | DNA/RNA | Short reads. Based on GCSA. Reference. |
| RNASTAR | RNA | Short reads. Extremely fast. High sensitive and accuracy. Based on Maximal Mappable Prefixes (MMPs). Reference. |
| BWA-MEM2 | DNA | Short reads. Twice as faster as BWA-MEM. Memory efficient. Based on Burrows-Wheeler. Reference. |
| Minimap2 | DNA/RNA | Long reads (PacBio and ONT). Extremely fast. Based on DALIGN and MHAP. Reference. |
| Bismark | DNA/RNA | Short reads. Bisulfite treated sequencing. Based on GCSA. Reference. |
| BBMap | DNA/RNA | Short and long reads (PacBio and ONT). Memory demanding. Reference. |
| Whisper 2 | DNA | Short reads. Indel sensitive. Variant-calling oriented. Reference. |
| S-conLSH | DNA | Long reads (ONT). High sensitivity and accuracy. Reference. |
File Formats
SAM/BAM file format

SAM: Sequence Alignment Map
BAM: Binary (compressed) SAM; not human-readable
SAM/BAM file format

- Original read information (from FASTQ) plus mapping information
- Position on reference, alignment, quality score, uniqueness, ..
Speaker Notes Alignment given in CIGAR string.
- in screenshot “37M” means 37 matches
- in screenshot “18M2D19M” means 18 matches, then 2 deletions, then 19 matches
class: top
Genome Browsers
- Visualise aligned reads (BAM files)
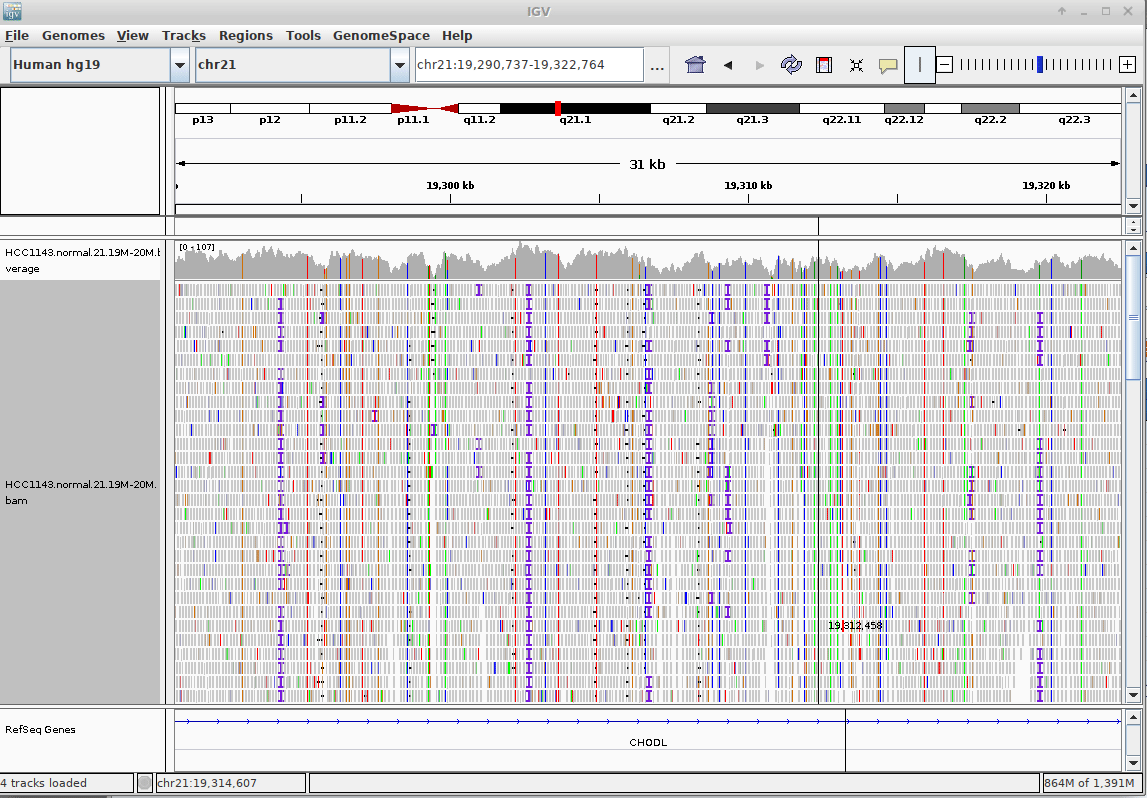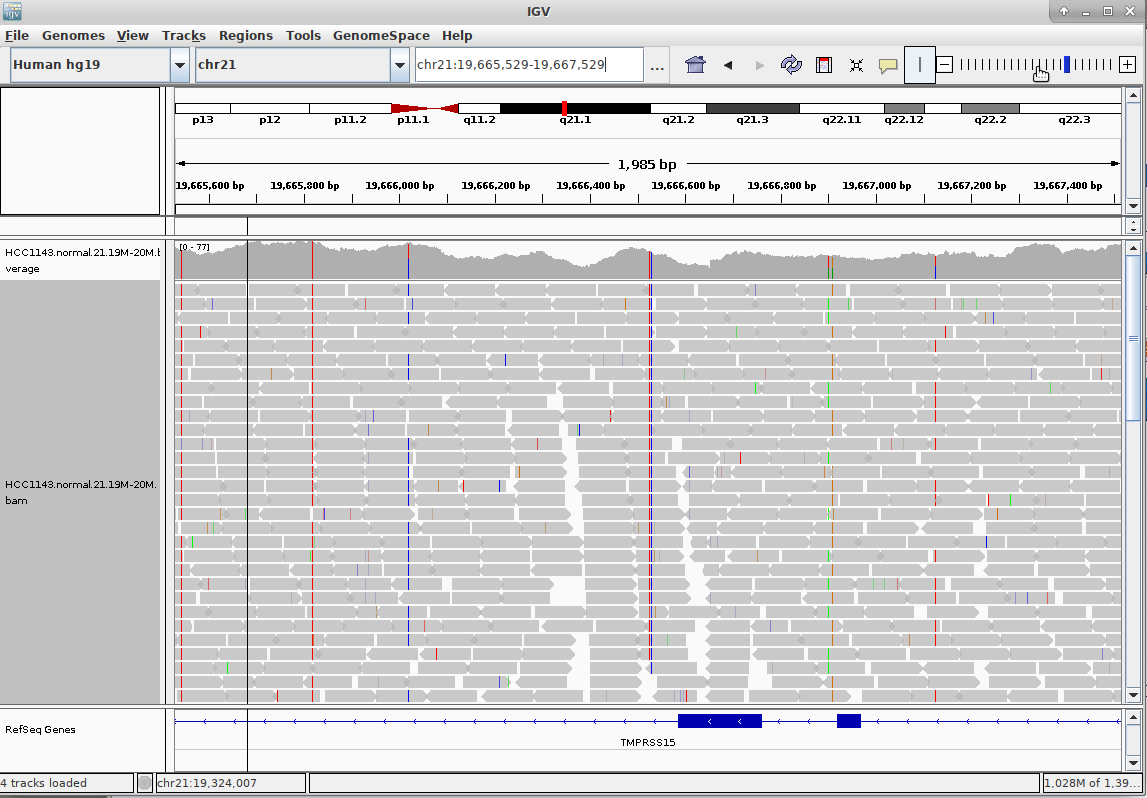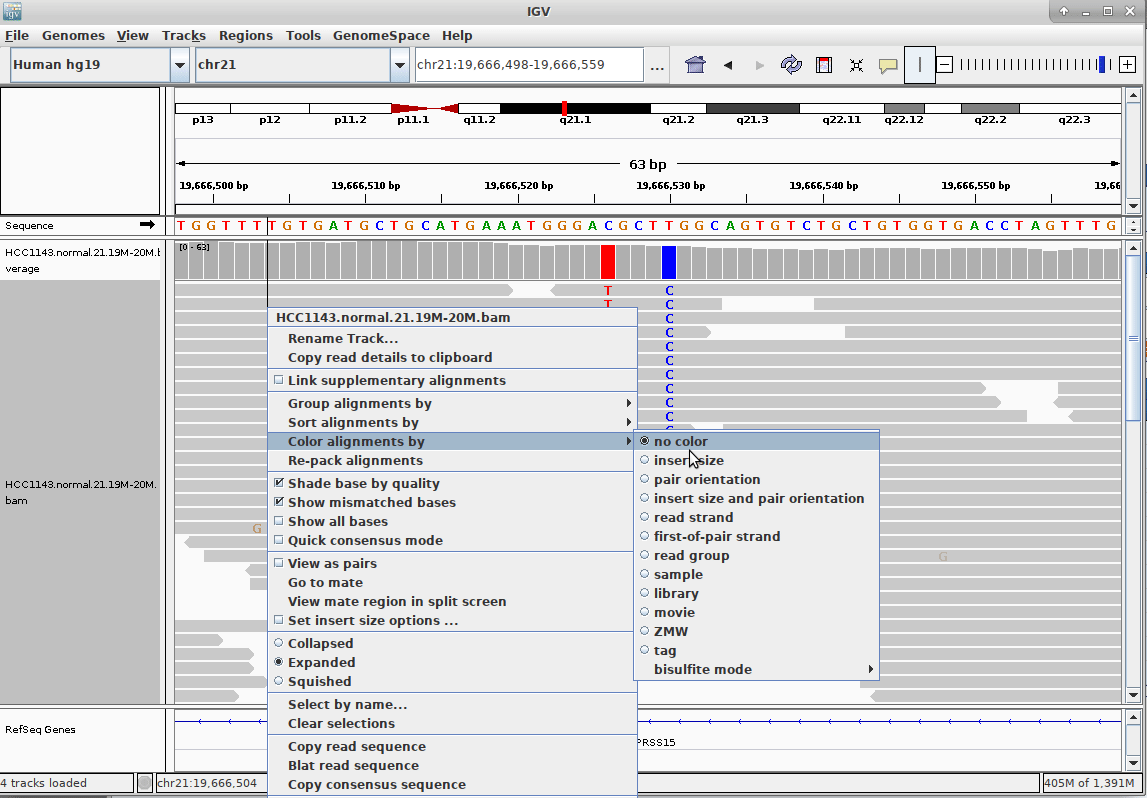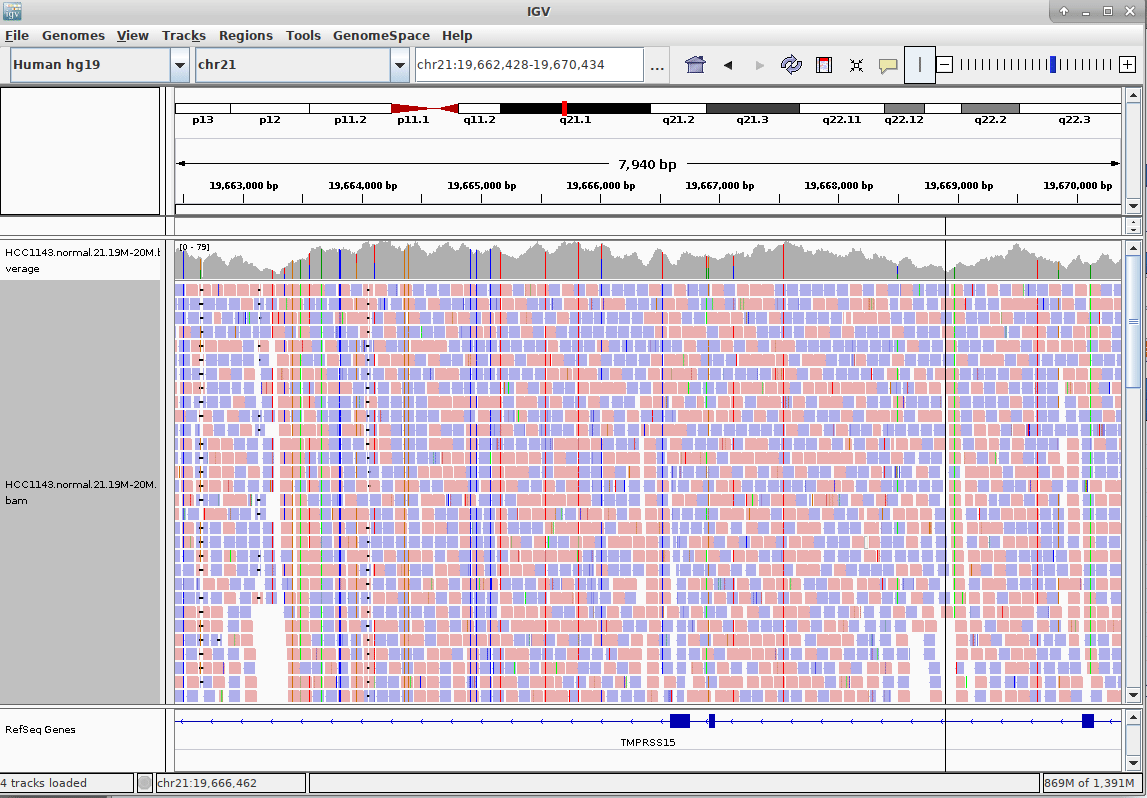
.footnote[This is IGV (Integrative Genome Browser) DOI: 10.1038/nbt.1754]
Speaker Notes
- Can zoom in and out, drag left and right, explore your sample
- Zoom in for more information, mismatches, read information
- Many different genome browsers exist
class: top
Genome Browsers in Galaxy
- JBrowse tool Genome Browser as Galaxy tool
.image-90[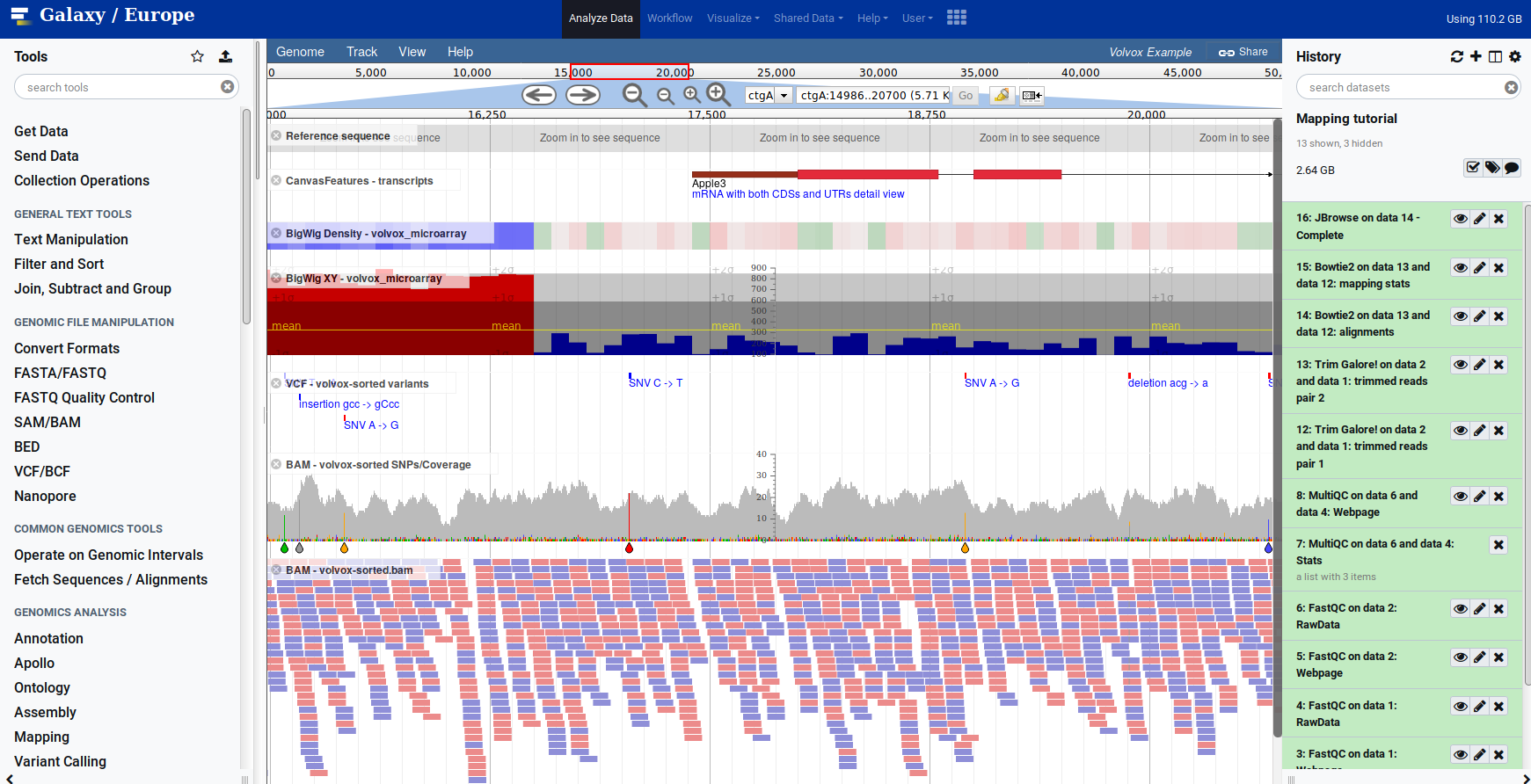 ]
]
.footnote[JBrowse.org DOI: 10.1186/s13059-016-0924-1]
Speaker Notes
Jbrowse tool builds up a small website for you, and pre-processes the reference genome into a more efficient format. If you wanted to share this with your colleagues, you could download this dataset and directly place it on your webserver.
class: top
Genome Browsers in Galaxy
- External Genome Browsers in Galaxy
- BAM datasets in Galaxy have display links
- UCSC Genome Browser, Ensemble, IGV, IGB, BAM.iobo
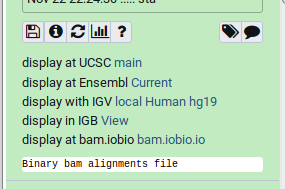
- Two different links for IGV
- local:
- Start IGV on your machine first
- Then click link to automatically load data from Galaxy
- [Reference genome name] (“Human hg19” in screenshot)
- Downloads and starts IGV for you
- Requires Java web start be installed on your machine
- local:
Speaker Notes In the mapping hands-on tutorial you will use JBrowse and IGV
Key Points
- Mapping is not trivial
- There are many mapping tools, best choice depends on your data
- Choice of mapper can affect downstream results
- Know your data!
- Genome browsers can be used to view aligned reads
Thank you!
This material is the result of a collaborative work. Thanks to the Galaxy Training Network and all the contributors! Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.
Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.
Funding
These individuals or organisations provided funding support for the development of this resource
