Bacterial Genome Annotation
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
Which genes are on a draft bacterial genome?
Which other genomic components can be found on a draft bacterial genome?
Requirements:
Run a series of tool to annotate a draft bacterial genome for different types of genomic components
Evaluate the annotation
Process the outputs to formate them for visualization needs
Visualize a draft bacterial genome and its annotations
Time estimation: 3 hoursLevel: Introductory IntroductorySupporting Materials:Published: Feb 1, 2024Last modification: Oct 15, 2024License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00403rating Rating: 5.0 (2 recent ratings, 7 all time)version Revision: 6
After sequencing and assembly, a genome can be annotated. It is an essential step to describe the genome.
Genome annotation consists in describing the structure and function of the components of the genome, by predicting, analyzing, and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome (structural annotation) and determines what those genes do (functional annotation).
To illustrate the process to annotate a bacterial genome, we take an assembly of a bacterial genome (KUN1163 sample) generated by following a bacterial genome assembly tutorial from data produced in “Complete Genome Sequences of Eight Methicillin-Resistant Staphylococcus aureus Strains Isolated from Patients in Japan” (Hikichi et al. 2019).
Methicillin-resistant Staphylococcus aureus (MRSA) is a major pathogen causing nosocomial infections, and the clinical manifestations of MRSA range from asymptomatic colonization of the nasal mucosa to soft tissue infection to fulminant invasive disease. Here, we report the complete genome sequences of eight MRSA strains isolated from patients in Japan.
AgendaIn this tutorial, we will cover:
Galaxy and data preparation
Any analysis should get its own Galaxy history. So let’s start by creating a new one and get the data (contig file from the assembly) into it.
Hands On: Prepare Galaxy and data
Create a new history for this analysis
To create a new history simply click the new-history icon at the top of the history panel:
Rename the history
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter
Import the contig file from Zenodo or from Galaxy shared data libraries:
https://zenodo.org/record/10572227/files/DRR187559_contigs.fasta
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import


Contig annotation
For annotating the contigs, several tools exists to do that: Prokka (Seemann 2014), Bakta (Schwengers et al. 2021), etc. Here, we use Bakta as recommended by
Torsten Seemann as the successor of Prokka.
Bakta is a tool for the rapid & standardized annotation of bacterial genomes and plasmids from both isolates and metagenome-assembled genomes (MAGs). It implements a comprehensive annotation workflow for coding and non-coding genes (i.e. tRNA, rRNA).
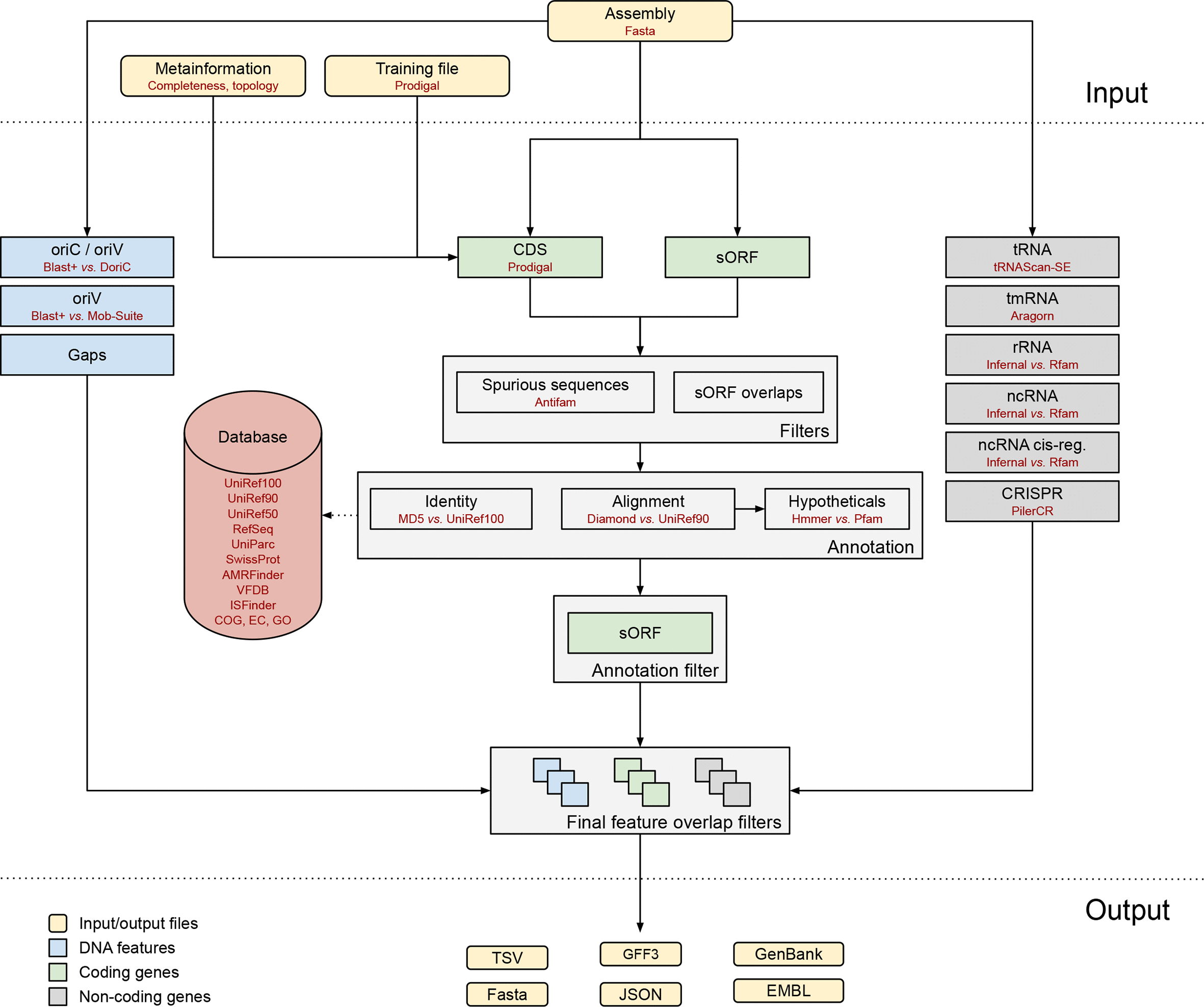 Open image in new tab
Open image in new tabIt is also able to detect and annotate small proteins (sORF). Predicted CDS are annotated using an alignment-free protein sequence identification approach with cross-references to public databases via stable identifiers.
Hands On: Contig annotation
- Bakta ( Galaxy version 1.9.3+galaxy0) with the following parameters:
- In “Input/Output options”:
- param-file “Select genome in fasta format”: Contig file
- “Bakta database”: latest one
- “AMRFinderPlus database”: latest one
- In “Optional annotation”:
- “Keep original contig header”:
Yes- In “Selection of the output files”:
- “Output files selection”:
Annotation file in TSVAnnotation and sequence in GFF3Feature nucleotide sequences as FASTASummary as TXTPlot of the annotation result as SVG
Bakta can generate many outputs. Here we selected:
-
Analysis_summary: a summary of the analysis as text fileQuestion- How many contigs have been provided as input?
- How long is the draft genome?
- How many CDSs have been found?
- How many small proteins?
- Which other components have been found?
How does it compare to results for KUN1163 in Table 1 in Hikichi et al. 2019?
- 44 (the sequence count)
- 2,911,349 bp, with is a bit shorter than the expected 2,914,567 bp in Table 1 in Hikichi et al. 2019
- 2,717 CDSs, a bit more than the expected 2,704 CDSs in Table 1 in Hikichi et al. 2019
- 5 sORFs. There is no information about sORFs in Hikichi et al. 2019
-
Other components
Components Bakta Hikichi et al. 2019 tRNAs 57 61 Transfer-messenger RNA (tmRNAs) 1 1 rRNAs 9 5 ncRNAs 95 No information
-
Nucleotide_sequenceswith feature nucleotide sequences as FASTA fileQuestion- How many sequences are in this file?
- What are the sequences stored there?
- 2,884 sequences
- Here are stored:
- tRNAs
- tmRNAs
- rRNAs
- ncRNAs
- CDSs
- sORFs
-
annotation_summarywith annotations as simple human readble TSVQuestionWhat is stored there?
This a table with 9 columns (
Sequence Id,Type,Start,Stop,Strand,Locus Tag,Gene,Product,DbXrefs). It contains here 2,916 lines, so the information and location for:- tRNAs
- tmRNAs
- rRNAs
- ncRNAs
- CDSs
- sORFs
- gaps
- oriCs
- oriVs
- oriTs
-
Annotation_and_sequencesin GFF3GFF is a file format used for describing genes and other features of DNA, RNA and protein sequences. It is a tab delimited file with 9 fields per line:
- seqid: The name of the sequence where the feature is located.
- source: The algorithm or procedure that generated the feature. This is typically the name of a software or database.
- type: The feature type name, like “gene” or “exon”. In a well structured GFF file, all the children features always follow their parents in a single block (so all exons of a transcript are put after their parent “transcript” feature line and before any other parent transcript line). In GFF3, all features and their relationships should be compatible with the standards released by the Sequence Ontology Project.
- start: Genomic start of the feature, with a 1-base offset. This is in contrast with other 0-offset half-open sequence formats, like BED.
- end: Genomic end of the feature, with a 1-base offset. This is the same end coordinate as it is in 0-offset half-open sequence formats, like BED.
- score: Numeric value that generally indicates the confidence of the source in the annotated feature. A value of “.” (a dot) is used to define a null value.
- strand: Single character that indicates the strand of the feature. This can be “+” (positive, or 5’->3’), “-“, (negative, or 3’->5’), “.” (undetermined), or “?” for features with relevant but unknown strands.
- phase: phase of CDS features; it can be either one of 0, 1, 2 (for CDS features) or “.” (for everything else). See the section below for a detailed explanation.
- attributes: A list of tag-value pairs separated by a semicolon with additional information about the feature.
QuestionHow many features are annotated?
51k+ (number of lines in the GFF)
-
Plot of the annotation as circular genome annotation
 Question
Question- What the 2 rings in the center?
- How are plotted the features?
- The first ring represents the GC content per sliding window over the entire sequence(s) with in green representing GC above and red GC below average. The 2nd ring represents the GC skew in orange and blue.
- All features are plotted on two rings representing the forward and reverse strand from outer to inner with CDS in grey (the other colors are hard to distinguish)
Further structural annotation
Bakta gives a lot of information already, especially regarding CDSs or RNAs, but some structural annotation might be missing, e.g. plasmids, or interesting to identify independently.
Plasmids
To identify plasmids in our contigs, we use PlasmidFinder (Carattoli and Hasman 2020), a tool for the identification and typing of plasmid sequences in Whole-Genome Sequencing. It uses the plasmidfinder database with hundreds of sequences to predict the plasmid in the data.
Hands On: Plasmid identification
- PlasmidFinder ( Galaxy version 2.1.6+galaxy1) with the following parameters:
- In “Input parameters”:
- param-file “Choose a fasta or fastq file”: Contig file
- “PlasmidFinder database”: most recent one
PlasmidFinder generates several outputs:
raw_results.txt: A text file containing the result table and alignments-
results.tsv: A tabular file with the following columns:- Database
- Plasmid: Plasmid against which the input genome has been aligned.
- Identity: Percent identity in the alignment between the best matching plasmid in the database and the corresponding sequence in the inputgenome (also called the high-scoring segment pair (HSP)). A perfect alignment is 100%, but must also cover the entire length of the plasmid in the database (compare example 1 and 3).
- Query/Template Length: Query length is the length of the best matching plasmid in the database, while HSP length is the length of the alignment between the best matching plasmid and the corresponding sequence in the genome (also called the high-scoring segment pair (HSP)).
- Contig: Name of contig the plasmid is found in.
- Position in contig: Starting position of the found gene in the contig.
- Note: Notes about the plasmid
- Accession number: Reference Genbank accession number accoding to NCBI for the plasmid in the database.
Question- How many plasmid sequences have been found?
- Where are they located?
- Are these sequences all associated with Staphylococcus aureus?
- What can we conclude about contig00019?
- 5 plasmid sequences
- 3 are on the contig00019, 1 on contig00002, and 1 on contig00024
- Looking at the accession number on the NCBI, we find that:
- CP000737, AP003139 (2 times) correspond to Staphylococcus aureus plasmids
- AF503772 corresponds to a Enterococcus faecalis plasmid
- CP003584 corresponds to a Enterococcus faecium plasmid
- All plasmid sequences corresponding to Staphylococcus aureus plasmids are all on contig00019, making this contig likely a plasmid. In addition, this contig has a length of 30,347 bp, which is similar to the expected length of the plasmid for KUN1163 in Table 1 in Hikichi et al. 2019
plasmid.fasta: A fasta file containing the best matching sequences from the query genomehit_in_genome.fasta: A fasta file containing the best matching plasmid genes from the database
Integrons
Integrons are genetic mechanisms that allow bacteria to adapt and evolve rapidly through the stockpiling and expression of new genes. An integron is minimally composed of:
- a gene encoding for a site-specific recombinase (intI)
- a proximal recombination site (attI), which is recognized by the integrase and at which gene cassettes may be inserted
- a promoter (Pc) which directs transcription of cassette-encoded genes
To detect integrons, we will use IntegronFinder (Néron et al. 2022). This tool:
- Annotates the CDS with Prodigal
-
Detects independently:
- integron integrase using the intersection of two HMM profiles: one specific of tyrosine-recombinase (PF00589) and one specific of the integron integrase, near the patch III domain of tyrosine recombinases
- attC recombination sites with a covariance model (CM), which models the secondary structure in addition to the few conserved sequence positions.
-
Integrates the results to distinguish 3 types of elements
- Complete integron: Integron with integron integrase nearby attC site(s)
- In0 element: Integron integrase only, without any attC site nearby
- CALIN element: Cluster of attC sites Lacking INtegrase nearby
Hands On: Integron identification
- IntegronFinder ( Galaxy version 2.0.5+galaxy0) with the following parameters:
- param-file “Replicon file”: Contig file
- “Thorough local detection”:
Yes- “Search also for promoter and attI sites?”:
Yes- “Remove log file”:
Yes
IntegronFinder generates 2 outputs:
-
A summary with for each sequence in the input the number of identified CALIN elements, In0 elements, and complete integrons.
QuestionHow many integron elements have been found?
No integron elements have been found on any contig. It could be because the genome is too stable, or because the assembly quality is not good enough and some parts useful for the integron detection were removed.
-
An integron annotation file as a tabular
IS (Insertion Sequence) elements
Insertion sequence (IS) element is a short DNA sequence that acts as a simple transposable element. IS are the smallest but most abundant autonomous transposable elements in bacterial genomes. They only code for proteins implicated in the transposition activity. They play then a key role in bacterial genome organization and evolution.
To detect IS elements, we will use ISEScan (Xie and Tang 2017). ISEScan is a highly sensitive software pipeline based on profile hidden Markov models constructed from manually curated IS elements
Hands On: IS identification
- ISEScan ( Galaxy version 1.7.2.3+galaxy1) with the following parameters:
- param-file “Genome fasta input”: Contig file
ISEScan generates several files:
-
A summary as a table
-
The results as a table
Question- How many IS elements have been detected?
- Where are they located?
- What the different IS families?
- 20
-
Using Group data by a column to group and count on 1st column, we find:
Contig IS element number contig00001 2 contig00002 1 contig00003 2 contig00004 1 contig00005 1 contig00006 1 contig00009 3 contig00010 1 contig00011 1 contig00012 1 contig00019 3 contig00027 1 contig00032 1 contig00037 1 -
As for previous question, when grouping and counting on 2nd column, we find 5 IS families:
IS families Identified IS elements IS1182 4 IS21 7 IS3 3 IS6 5 ISL3 1
- The results as a GFF file
- Several FASTA files:
- IS nucleotide sequences
- ORF nucleotide sequences
- ORF amino acide sequences
Visualisation of the annotation
We would like to look at the annotation using JBrowse (Diesh et al. 2023) with several information:
- Annotations identified by Bakta
- Plasmid sequences identified by PlasmidFinder
- Integrons identified by IntegronFinder
- IS elements identified by ISEscan
JBrowse needs the annotations to be in GFF format. Bakta and ISEscan generated both GFF files. For PlasmidFinder and IntegronFinder, we need to format the outputs.
PlasmidFinder generated the results.tsv with all needed information. To transform it to a GFF, we need to:
- Split the 6th column on
..to have start and end into 2 separated columns - Remove in the content of column 5 what is after the contig name
- Remove the 1st line
- Transform to GFF3
Hands On: Transform PlasmidFinder to GFF
- Replace Text in a specific column ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “File to process”:
results.tsvoutput of PlasmidFinder- In “Replacement”:
- In “1: Replacement”
- in column:
Column: 6- “Find pattern”:
(.*)\.\.(.*)- “Replace with”:
\\1\t\\2This will split the content of the 6th column on
..and put it into column 6 and column 7. Column 7 will be then replaced.- param-repeat “Insert Factor”
- In “2: Replacement”
- in column:
Column: 5- “Find pattern”:
(.*)( len.*)- “Replace with”:
\\1This will remove in the content of column 5 what is after the contig name
- Select last lines from a dataset ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “Text file”: output of Replace Text above
- “Operation”:
Keep everything from this line on- “Number of lines”:
2- Table to GFF3 ( Galaxy version 1.2) with the following parameters:
- param-file “Table”: output of the above Select last tool step
- “Record ID column or value”:
5- “Start column or value”:
6- “End column or value”:
7- “Type column or value”:
2- “Score column or value”:
3- “Source column or value”:
1- param-repeat “Insert Qualifiers”
- “Name”:
name- “Qualifier value column or raw text”:
8- param-repeat “Insert Qualifiers”
- “Name”:
accession- “Qualifier value column or raw text”:
9- Rename to
PlasmidFinder GFF
IntegronFinder tabular output can be transformed to GFF by:
- Replace
NAvalues on column 7 by0- Remove the first two lines
- Transform to GFF3
Hands On: Transform IntegronFinder to GFF
- Replace Text in a specific column ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “File to process”: tabular output of IntegronFinder
- In “Replacement”:
- In “1: Replacement”
- in column:
Column: 7- “Find pattern”:
NA- “Replace with”:
0- Select last lines from a dataset ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “Text file”: output of Replace Text above
- “Operation”:
Keep everything from this line on- “Number of lines”:
3- Table to GFF3 ( Galaxy version 1.2) with the following parameters:
- param-file “Table”: tabular output of IntegronFinder
- “Record ID column or value”:
2- “Start column or value”:
4- “End column or value”:
5- “Type column or value”:
11- “Score column or value”:
7- “Source column or value”:
IntegronFinder- param-repeat “Insert Qualifiers”
- “Name”:
name- “Qualifier value column or raw text”:
3- param-repeat “Insert Qualifiers”
- “Name”:
annotation- “Qualifier value column or raw text”:
9- Rename to
IntegronFinder GFF
We can now launch JBrowse with different information track.
Hands On: Visualize the Genome
- JBrowse ( Galaxy version 1.16.11+galaxy1) with the following parameters:
- “Reference genome to display”:
Use a genome from history
- param-file “Select the reference genome”: Contig file
- “Genetic Code”:
11. The Bacterial, Archaeal and Plant Plastid Code- In “Track Group”:
- param-repeat “Insert Track Group”
- “Track Category”:
Bakta- In “Annotation Track”:
- param-repeat “Insert Annotation Track”
- “Track Type”:
GFF/GFF3/BED Features
- param-file “GFF/GFF3/BED Track Data”:
Annotation_and_sequences(output of Bakta tool)- “JBrowse Track Type [Advanced]”:
Neat Canvas Features- “Track Visibility”:
On for new users- param-repeat “Insert Track Group”
- “Track Category”:
Plasmid sequences- In “Annotation Track”:
- param-repeat “Insert Annotation Track”
- “Track Type”:
GFF/GFF3/BED Features
- param-file “GFF/GFF3/BED Track Data”:
PlasmidFinder GFF- “JBrowse Track Type [Advanced]”:
Neat Canvas Features- “Track Visibility”:
On for new users- param-repeat “Insert Track Group”
- “Track Category”:
IS elements- In “Annotation Track”:
- param-repeat “Insert Annotation Track”
- “Track Type”:
GFF/GFF3/BED Features
- param-file “GFF/GFF3/BED Track Data”: GFF output of ISEScan
- “JBrowse Track Type [Advanced]”:
Neat Canvas Features- “Track Visibility”:
On for new usersIf integrons are found as IntegronFinder
- param-repeat “Insert Track Group”
- “Track Category”:
Integrons- In “Annotation Track”:
- param-repeat “Insert Annotation Track”
- “Track Type”:
GFF/GFF3/BED Features
- param-file “GFF/GFF3/BED Track Data”:
IntegronFinder GFF- “JBrowse Track Type [Advanced]”:
Neat Canvas Features- “Track Visibility”:
On for new users- View the output of JBrowse
In the output of the JBrowse you can view the genes, IS, plasmid, etc on the contigs. With the search tools you can easily find genes of interest. JBrowse can handle many inputs and can be very useful.
If it takes too long to build the JBrowse instance, you can view an embedded one here:
Comment
- It is ok to have the message stating
Error reading from name store..- The feature name search will not work.
Question
- Have all sequences identified by PlasmidFinder on contig19 been identified by Bakta?
- Have all sequences identified by ISEScan on contig19 been identified by Bakta?
- Yes all sequences in the PlasmidFinder track are also in the Bakta track. For
- All Insertion Sequences in the ISEScan track are also in the Bakta track, but the Terminanl Inverted repeats are not in the Bakta track
To learn more options for JBrowse, check out the dedicated tutorial
Conclusion
In this tutorial, contigs were annotated with different tools and then visualized.
Other visualizations, specially for publications, can be done using Circos. To learn how to do it, you can follow the dedicated tutorial.
To refine the genome annotation, you can also use Apollo and our tutorial.
You've Finished the Tutorial
Key points
Bakta is a powerful tool to annotate a bacterial genome
Annotation can be easily visualized to understand the genomic context and help making sense of the annotations
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsReferences
- Seemann, T., 2014 Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. 10.1093/bioinformatics/btu153
- Xie, Z., and H. Tang, 2017 ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33: 3340–3347. 10.1093/bioinformatics/btx433
- Hikichi, M., M. Nagao, K. Murase, C. Aikawa, T. Nozawa et al., 2019 Complete Genome Sequences of Eight Methicillin-Resistant Staphylococcus aureus Strains Isolated from Patients in Japan (I. L. G. Newton, Ed.). Microbiology Resource Announcements 8: 10.1128/mra.01212-19
- Carattoli, A., and H. Hasman, 2020 PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Horizontal gene transfer: methods and protocols 285–294. 10.1007/978-1-4939-9877-7_20
- Schwengers, O., L. Jelonek, M. A. Dieckmann, S. Beyvers, J. Blom et al., 2021 Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microbial genomics 7: 000685. 10.1099/mgen.0.000685
- Néron, B., E. Littner, M. Haudiquet, A. Perrin, J. Cury et al., 2022 IntegronFinder 2.0: identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 10: 700. 10.3390/microorganisms10040700
- Diesh, C., G. J. Stevens, P. Xie, T. De Jesus Martinez, E. A. Hershberg et al., 2023 JBrowse 2: a modular genome browser with views of synteny and structural variation. Genome biology 24: 1–21. 10.1186/s13059-023-02914-z
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Bérénice Batut, Bacterial Genome Annotation (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/bacterial-genome-annotation/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{genome-annotation-bacterial-genome-annotation, author = "Bérénice Batut", title = "Bacterial Genome Annotation (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/bacterial-genome-annotation/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!


Do you want to extend your knowledge?Follow one of our recommended follow-up trainings:
- slides Slides: Refining Genome Annotations with Apollo (prokaryotes)
- tutorial Hands-on: Refining Genome Annotations with Apollo (prokaryotes)
- slides Slides: Visualisation with Circos
- tutorial Hands-on: Visualisation with Circos
- slides Slides: Genomic Data Visualisation with JBrowse
- tutorial Hands-on: Genomic Data Visualisation with JBrowse
- tutorial Hands-on: Extracting Workflows from Histories
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/genome-annotation/tutorials/bacterial-genome-annotation/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: 86755160afbf tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: 86755160afbf tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bakta owner: iuc revisions: ba6990f72184 tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: integron_finder owner: iuc revisions: bfd290fe1588 tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: isescan owner: iuc revisions: 9e776e7fab4f tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: jbrowse owner: iuc revisions: a6e57ff585c0 tool_panel_section_label: Graph/Display Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: plasmidfinder owner: iuc revisions: 7075b7a5441b tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: tbl2gff3 owner: iuc revisions: 4a7f4b0cc0a3 tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/