Frequently Asked Questions
Tutorial Questions
Is it possible to replace the existing alignment tools such as HISAT and Freebayes with other tools?
The tools in this workflow are customizable, however, the user has to ensure that the inputs are in the correct format, while using the same reference genome database.
Why do we change the chromosome names in the Ensembl GTF to match the UCSC genome reference?
UCSC chromosome names begin with the prefix
chr, but Ensembl chromosome names do not. For example, chromosome 19 would be denoted aschr19in UCSC, and as19in Ensemble. Most tools would view those as different when looking for matches/overlaps. Therefore it is always a good idea to make sure these match before you perform any downstream analysis.
Why do we have a variant mapping file when it is not being used in the workflow?
We are working on updating the existing annotation tool to include the variant mapping file. Once that is done, the variant mapping file will also be an input for those tools.
Proteogenomics general
Can I use these workflows on datasets generated from our laboratory?
Yes, the workflows can be used on other datasets as well. However, you will need to consider data acquisition and sample preparation methods so that the tool parameters can be adjusted accordingly.
Example histories for the proteogenomics tutorials
If you get stuck or would like to see what the results should look like, you can have a look at one of the following public histories:
Galaxy EU (usegalaxy.eu):
- History1: Part 1: database creation
- History2: Part 2: database search
- History3: Part3: novel peptide analysis
Galaxy Main (usegalaxy.org):
- History 1: Part 1: database creation
- History 2: Part 2: database search
- History 3: Part 3: novel peptide analysis
The workflows contain several Query tabular for text manipulation, is there a tutorial for that?
Query tabular loads a tabular database and creates a sqlite database and tabular file. To learn more about SQL Queries - please look at this documentation.
The help section on the Query Tabular tool provides simple examples of both filtering the input tabular datasets, as well as examples of SQL queries. Query Tabular also incorporates regex functions that can be used queries. The PSM report datasets in these tutorials have fields that are lists of protein IDs.
Query Tabular help shows how to normalize those protein list fields so that we can perform operations by protein ID. See section: Normalizing by Line Filtering into 2 Tables in the tool help (below the tool in Galaxy).
What kind of variants are seen in the output?
From this workflow we can see insertions, deletions, SNVs, or we will know whether it’s an intron, exon, splice junction etc.
Visualisation
Using IGV with Galaxy
You can send data from your Galaxy history to IGV for viewing as follows:
- Install IGV on your computer (IGV download page)
- Start IGV
- In recent versions of IGV, you will have to enable the port:
- In IGV, go to
View > Preferences > Advanced- Check the box
Enable Port- In Galaxy, expand the dataset you would like to view in IGV
- Make sure you have set a reference genome/database correctly (dbkey) (instructions)
- Under
display in IGV, click onlocal
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
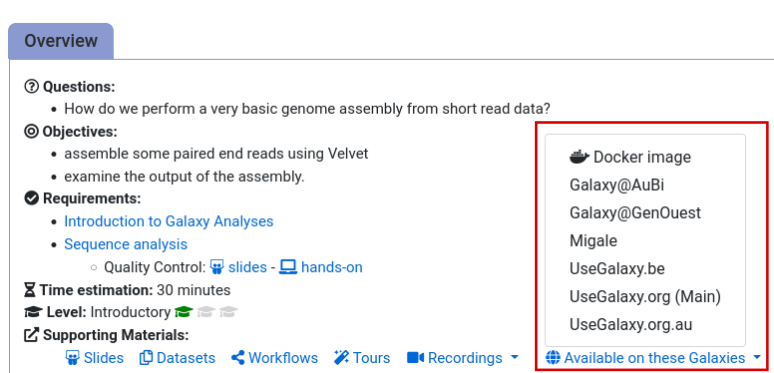
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history