Predicting EI+ mass spectra with QCxMS
Contributors
last_modification Published: Oct 1, 2024
last_modification Last Updated: Oct 1, 2024
qcxms-spectra-predictions
Motivation
.pull-left[
- MS data annotation poses a universal bottleneck in research.
- In silico spectra prediction using machine learning or quantum chemistry is a promising technique for annotation of unknown compounds.
- QCxMS offers reasonably accurate in silico annotation, especially for organic molecules.
- The complexity of quantum chemistry predictions presents challenges for non-HPC experts.
- Integrating QCxMS into Galaxy provides valuable molecular insights.
Our Goal: Make semi-empirical Quantum Chemistry (QC)-based predictions accessible without advanced computational skills. ]
.pull-right[
 ]
]
Method
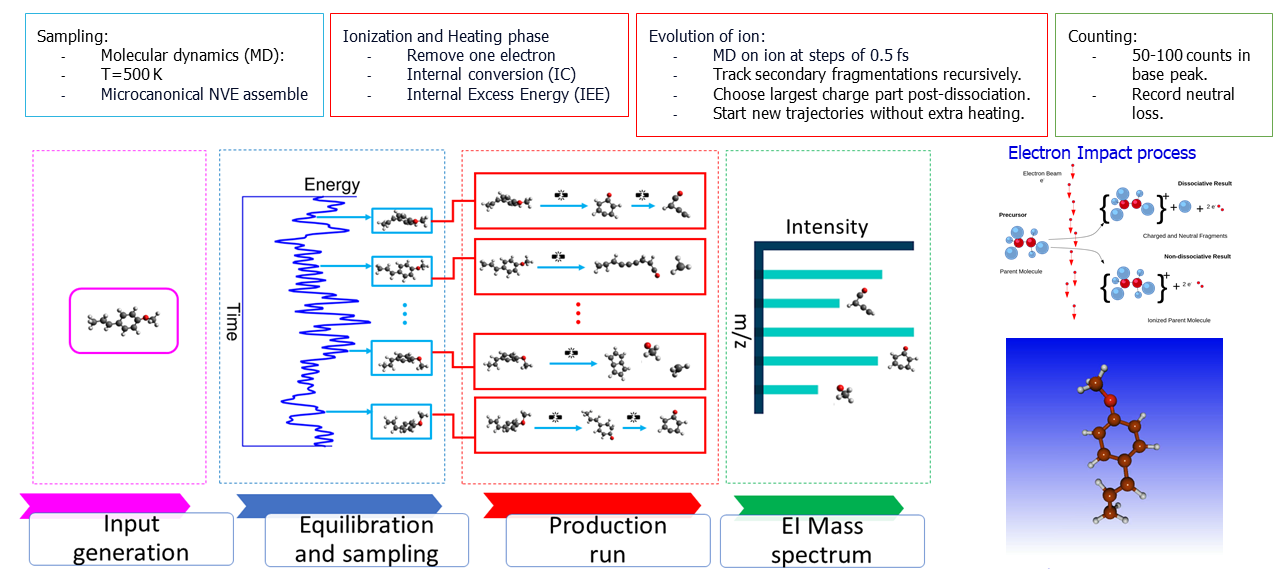
- Calculations can be performed using multiple quantum chemistry frameworks.
- This includes full ab initio molecular dynamics or parameterized semi-empirical quantum mechanical methods.
- Semi-empirical methods are significantly faster (hours vs. weeks) while yielding reasonable accuracy.
HPC Workflow
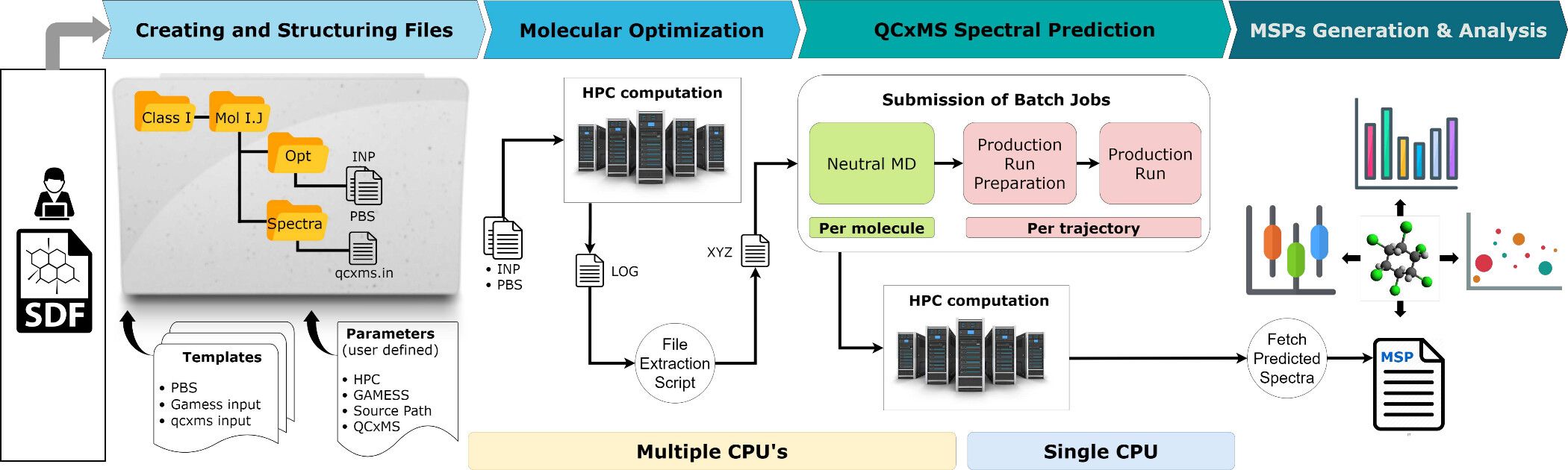
- The HPC-based workflow is configured to work with a PBS job scheduler and is therefore somwhat limited in flexibility and user friendliness.
- Due to the difficulties of working with HPC clusters, we ported this workflow to Galaxy.
Galaxy Workflow
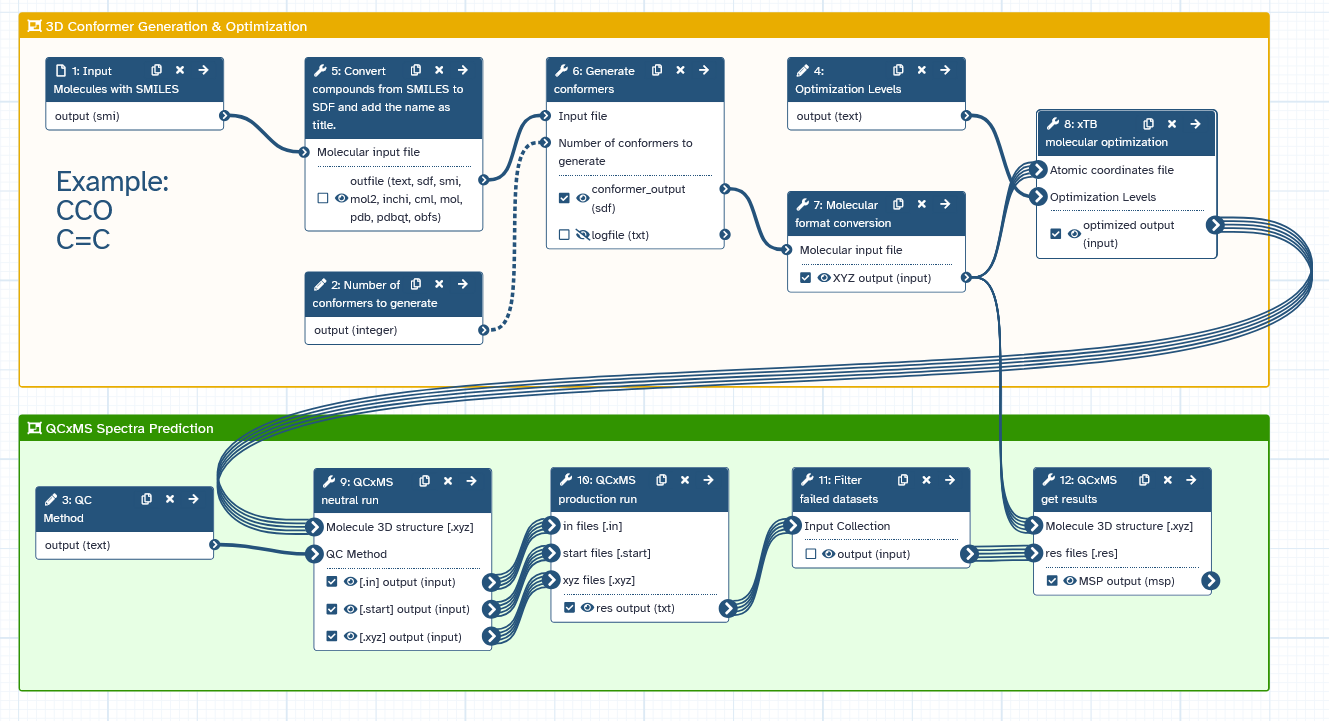
- We use xTB to optimize the input geometry.
- The QCxMS neutral run tool performs the equilibration and sampling to create the trajectories.
- The QCxMS production run tool performs the simulation of each trajectory.
- The QCxMS getres tool collects the results into an MSP file.
Galaxy Tool Structure
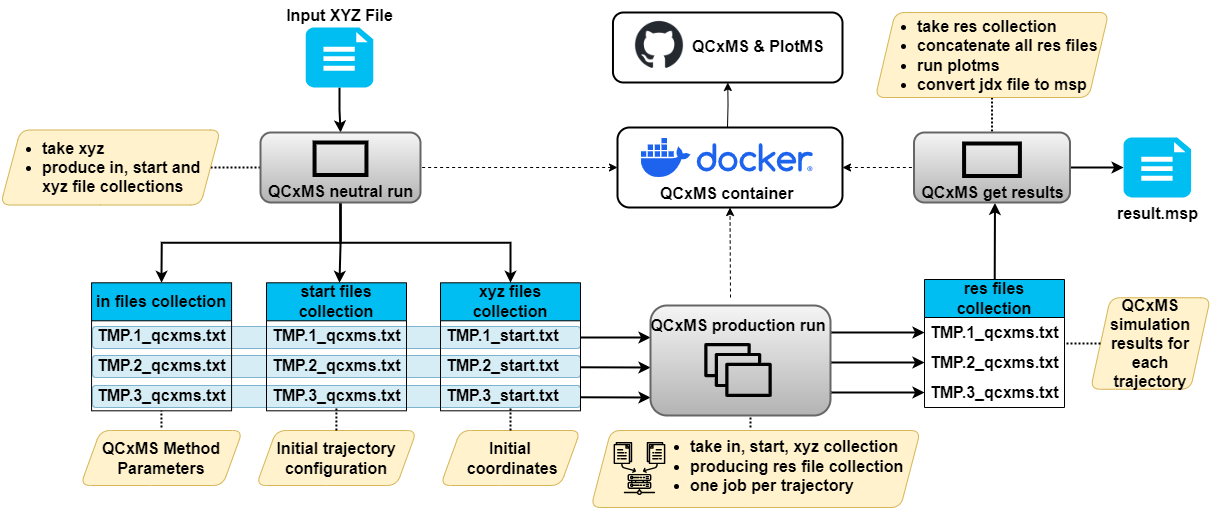
- Each trajectory is computed in a single job, using data-level parallelism.
- All QCxMS tools utilize the same docker container with an executable which was compiled with an optimized compiler.
Runtime Performance Metrics
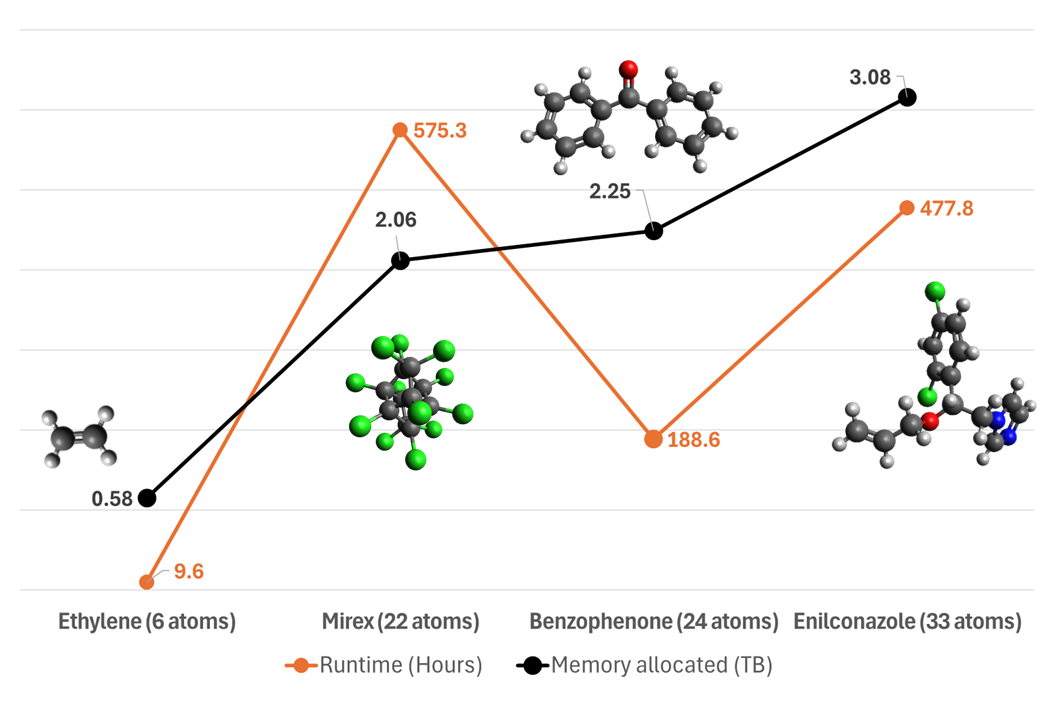
- Runtime depends primarily on size of the molecule.
- Structure also plays a crucial role - the more complex the geometry and structure, the longer the computations take.
- Memory requirements scale linear with size of the molecule.
References
- Grimme, S. (2013). Towards First Principles Calculation of Electron Impact Mass Spectra of Molecules. Angewandte Chemie International Edition, 52(24), 6306–6312. https://doi.org/10.1002/anie.201300158
- Bauer, C. A., & Grimme, S. (2016). How to Compute Electron Ionization Mass Spectra from First Principles. The Journal of Physical Chemistry A, 120(21), 3755–3766. https://doi.org/10.1021/acs.jpca.6b02907
- Bannwarth, C., Ehlert, S., & Grimme, S. (2019). GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. Journal of Chemical Theory and Computation, 15(3), 1652–1671. https://doi.org/10.1021/acs.jctc.8b01176
- Koopman, J., & Grimme, S. (2021). From QCEIMS to QCxMS: A Tool to Routinely Calculate CID Mass Spectra Using Molecular Dynamics. Journal of the American Society for Mass Spectrometry, 32(7), 1735–1751. https://doi.org/10.1021/jasms.1c00098
- Hecht, H., Rojas, W. Y., Ahmad, Z., Křenek, A., Klánová, J., & Price, E. J. (2024). Quantum Chemistry-Based Prediction of Electron Ionization Mass Spectra for Environmental Chemicals. Analytical Chemistry, 96(33), 13652–13662. https://doi.org/10.1021/acs.analchem.4c02589
Thank you!
This material is the result of a collaborative work. Thanks to the Galaxy Training Network and all the contributors! Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.
Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.