Assembly of the mitochondrial genome from PacBio HiFi reads
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How to assemble the mitochondrial genome from PacBio Hifi Reads
Requirements:
Generate Mitochondrial assembly
Understand the outputs of MitoHifi
Time estimation: 1 hourSupporting Materials:Published: Sep 3, 2024Last modification: Apr 22, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00453rating Rating: 4.6 (0 recent ratings, 9 all time)version Revision: 5
This tutorial will show you how to assemble a mitochondrial genome from PacBio HiFi data using MitoHiFi Uliano-Silva et al. 2023. Combined with the tutorials “Using the VGP workflows to assemble a vertebrate genome with HiFi and Hi-C data” and “Decontamination of a genome assembly”, this allows you to produce a reference assembly for both the nuclear and the mitochondrial DNA of a vertebrate species.
This tutorial uses data from the Zebra Finch (Taeniopygia guttata) generated by the Vertebrate Genome Project. We downsampled the reads that didn’t align with the mitochondrial genome so that the tutorial can run faster.
Comment: Run this analysis on "real" dataIf you want to run this analysis on a real sequencing library generated by the Vertebrate Genome Project you can find the PacBio HiFi data on Genome Ark as a remote repository and upload it to Galaxy (available on the three main Public Galaxy instances: .org, .eu, .org.au).
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a Choose from repositories:
- Click on Upload Data on the top of the left panel
Click on Choose from repository and scroll down to find your repository or type the repository name in the
search boxon the top.
- Look for your data under:
Genome Ark -> species -> Taeniopygia_guttata -> bTaeGut2 -> genomic_data -> pacbio_hifi -> *_reads.fasta.gz- Select the datasets you want to import
- click on OK
- Click on Start
- Click on Close
- You can find the dataset has begun loading in you history.
The assembly is using the wrapped workflow MitoHiFi. MitoHiFi:
- Extracts mitochondrial reads (based on a BLAST against an existing reference mitogenome) and uses Hifiasm Cheng et al. 2021 to assemble them.
- Removes nuclear mitochondrial DNA sequences (NUMTs) from the potential mitogenome contigs
- Generates a circularized and annotated genome for all potential mitogenome contigs
- Selects a representative for the final mitochondrial assembly
AgendaIn this tutorial, we will cover:
Get data
Hands On: Data Upload
- Create a new history for this tutorial
Import the files from Zenodo:
https://zenodo.org/records/13345315/files/PacBio_reads.fastqsanger.gz
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
Check that the datatype is
fastqsanger.gz
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
fastqsanger.gzfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button

Download the mitogenome for a related species
To assemble the mitogenome from our PacBio Data, MitoHiFi needs a reference mitogenome to fish out mitochondrial reads (the reads are blasted against the related reference). To download this reference, we use the tool MitoHiFi with the operation Find a closely related species.
- MitoHiFi ( Galaxy version 3.2.3+galaxy0) with the following parameters:
- “Operation type selector”:
Find a close-related mitochondrial reference genome
- “Species name”:
Taeniopygia guttataEnter the latin name of the species you are assembling- “Email”:
your.email@service.comEnter your email- “Minimal appropriate length”:
15000As vertebrate mitochondrial genomes are typically at least 14kbp long, we are using a value in this range so that we get complete mitogenome results as our reference.
Assemble the mitochondrial genome with MitoHiFi
Hands On: Assemble the mitochondrial genome
Create a collection with your PacBio HiFi Reads
- Click on galaxy-selector Select Items at the top of the history panel
- Check the fastq.gz containing the HiFi reads
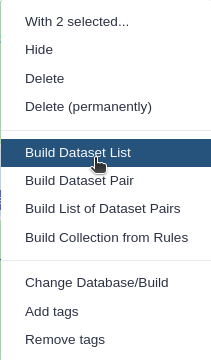
Click 1 of N selected and choose Advanced Build List
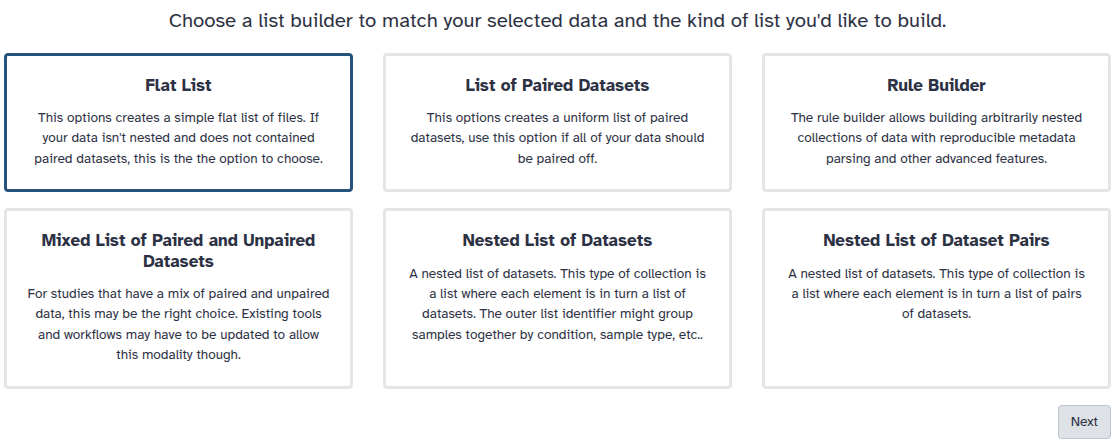
You are in collection building wizard. Choose Flat List and click ‘Next’ button at the right bottom corner.
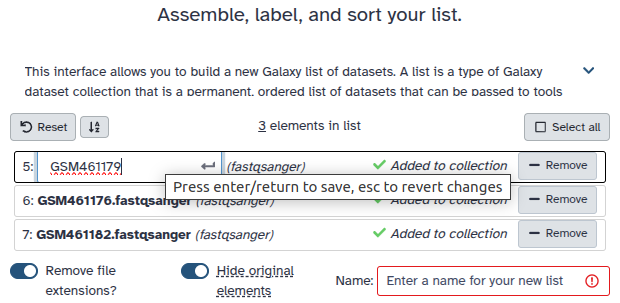
Double clcik on the file names to edit. For example, remove file extensions or common prefix/suffixes to cleanup the names.
- Enter a name for your collection to PacBio Reads
- Click Build to build your collection
- Click on the checkmark icon at the top of your history again
MitoHiFi ( Galaxy version 3.2.3+galaxy0) with the following parameters:
- “Operation type selector”:
Run MitoHiFi
- “Input mode”:
Pacbio Hifi Reads
- param-collection “Pacbio Hifi reads”:
PacBio Reads(Input dataset collection)- param-file “Close-related mitogenome in fasta format”:
MitoHiFi on : reference genome (FASTA)(output of MitoHiFi tool)- param-file “Close-related mitogenome in genbank format”:
MitoHiFi on : reference genome (genbank)(output of MitoHiFi tool)- “Genetic code”:
Vertebrate mitochondrial code- In “Advanced options”:
- “Blast percentage identity”:
70This setting filters the potential mito contigs – setting it to 70 means that we are retaining contigs with at least 70% of its length in the BLAST match. This parameter can be lowered if you are expecting more sequence divergence among mitogenomes of your taxa, or vice versa.
Outputs of MitoHiFi:
- Final mitogenome (FASTA). The mitochondrial genome circularized and rotated to start at tRNA-Phe.
- Final mitogenome (genbank). The final mitogenome annotated in GenBank format.
- Final mitogenome annotation (png). The predicted genes in the final mitogenome.
- Final mitogenome coverage (png). The sequencing coverage along the final mitogenome.
- Contigs stats (TSV). Contains the statistics of your assembled mitogenomes such as the number of genes, size, whether it was circularized or not, if the sequence has frameshifts, and other metrics.
- Reads mapped to close-related mtDNA (FASTA). All and filtered by size.
- Hifiasm contigs (fasta). The results of running Hifiasm on the mitochondrial reads.
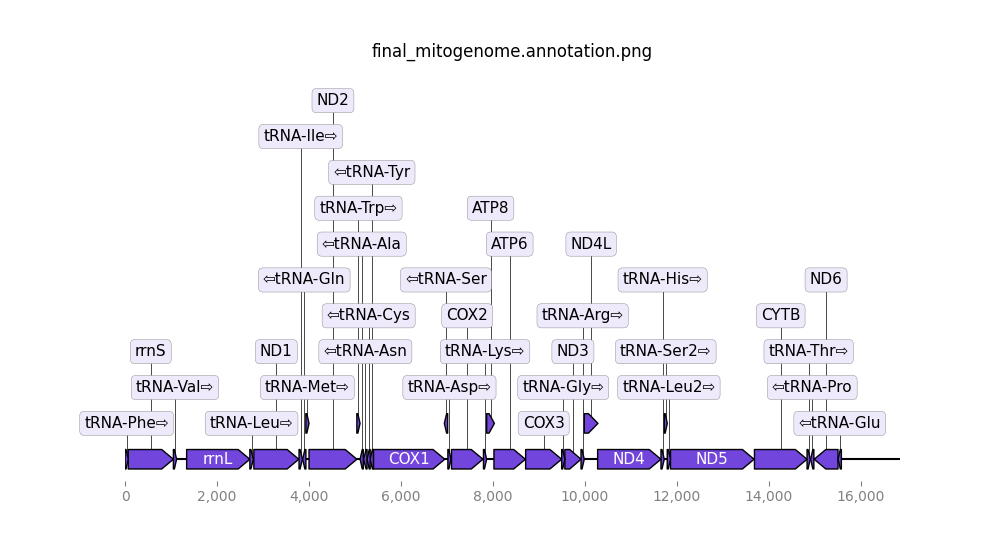 Open image in new tab
Open image in new tabConclusion
In this tutorial, we learned how to assemble the mitochondrial genome using PacBio HiFi reads and MitoHiFi. You can try this tutorial on your own data using the full HiFi read set you’d use for the nuclear genome assembly, since the filtering for mitochondrial reads happens within MitoHiFi.
You've Finished the Tutorial
Key points
MitoHifi generates the assembly and annotation of the Mitochondrial Genome using a reference sequence.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsReferences
- Cheng, H., G. T. Concepcion, X. Feng, H. Zhang, and H. Li, 2021 Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18: 170–175. 10.1038/s41592-020-01056-5
- Uliano-Silva, M., J. G. R. N. Ferreira, K. Krasheninnikova, G. Formenti, L. Abueg et al., 2023 MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC bioinformatics 24: 288. 10.1186/s12859-023-05385-y
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Delphine Lariviere, Assembly of the mitochondrial genome from PacBio HiFi reads (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/assembly/tutorials/mitochondrion-assembly/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{assembly-mitochondrion-assembly, author = "Delphine Lariviere", title = "Assembly of the mitochondrial genome from PacBio HiFi reads (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/assembly/tutorials/mitochondrion-assembly/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/assembly/tutorials/mitochondrion-assembly/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: mitohifi owner: bgruening revisions: bfab6f5b449d tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: compress_file owner: iuc revisions: '09ea79f9f260' tool_panel_section_label: Convert Formats tool_shed_url: https://toolshed.g2.bx.psu.edu/