Bei der Sequenzierung werden die Nukleotidbasen in einer DNA- oder RNA-Probe (Bibliothek) durch den Sequenzierer bestimmt. Für jedes Fragment in der Bibliothek wird eine Sequenz erzeugt, auch Read genannt, die einfach eine Folge von Nukleotiden ist.
Moderne Sequenzierungstechnologien können in einem einzigen Experiment eine riesige Anzahl von Sequenzier-Reads erzeugen. Allerdings ist keine Sequenzierungstechnologie perfekt, und jedes Gerät erzeugt unterschiedliche Arten und Mengen von Fehlern, wie z. B. falsch bezeichnete Nukleotide. Diese falsch benannten Basen sind auf die technischen Beschränkungen der einzelnen Sequenzierplattformen zurückzuführen.
Daher ist es notwendig, Fehlertypen zu verstehen, zu identifizieren und auszuschließen, die die Interpretation der nachgeschalteten Analyse beeinträchtigen können. Die Qualitätskontrolle der Sequenzen ist daher ein wesentlicher erster Schritt in Ihrer Analyse. Frühzeitiges Erkennen von Fehlern spart später Zeit.
Agenda
In diesem Tutorium werden wir uns mit folgenden Themen beschäftigen:
Erstellen Sie einen neuen Verlauf für dieses Tutorial und geben Sie ihm einen passenden Namen
Um einen neuen Verlauf zu erstellen, klicken Sie einfach auf das Symbol new-history am oberen Rand des Verlaufsfensters:
Klicken Sie auf galaxy-pencil (Bearbeiten) neben dem Namen der Geschichte (der standardmäßig “Unbenannte Geschichte” lautet)
Geben Sie den neuen Namen ein
Klicken Sie auf Speichern
Um die Umbenennung abzubrechen, klicken Sie auf die galaxy-undo schaltfläche “Abbrechen”
Wenn Sie nicht das galaxy-pencil (Edit) neben dem Verlaufsnamen haben (was der Fall sein kann, wenn Sie eine ältere Version von Galaxy verwenden), gehen Sie wie folgt vor:
Klicken Sie auf Unbenannter Verlauf (oder den aktuellen Namen des Verlaufs) (Klicken Sie zum Umbenennen des Verlaufs) oben in Ihrem Verlaufsfenster
Geben Sie den neuen Namen ein
Drücken Sie Enter
Importieren Sie die Datei female_oral2.fastq-4143.gz von Zenodo oder aus der Datenbibliothek (fragen Sie Ihren Dozenten) Dies ist eine Mikrobiomprobe von einer Schlange Jacques et al. 2021.
Klicken Sie auf galaxy-uploadDaten hochladen am oberen Rand der Werkzeugleiste
Wählen Sie galaxy-wf-editDaten einfügen/holen
Fügen Sie den/die Link(s) in das Textfeld ein
Drücken Sie Start
Schließen Sie das Fenster
Als Alternative zum Hochladen der Daten von einer URL oder Ihrem Computer können die Dateien auch von einer Shared Data Library zur Verfügung gestellt werden:
Gehen Sie in Bibliotheken (linker Bereich)
Navigieren Sie zu dem richtigen Ordner, wie von Ihrem Ausbilder angegeben.
Auf den meisten Galaxies werden die Tutoriumsdaten in einem Ordner mit dem Namen GTN - Material –> Topic Name -> Tutorial Name bereitgestellt.
Wählen Sie die gewünschten Dateien aus
Klicken Sie auf Zur Historie hinzufügengalaxy-dropdown am oberen Rand und wählen Sie as Datasets aus dem Dropdown-Menü
Wählen Sie im Pop-up-Fenster
“Historie auswählen “: die Historie, in die Sie die Daten importieren möchten (oder erstellen Sie eine neue)
Klicken Sie auf Importieren
Benennen Sie den importierten Datensatz in Reads um.
Wir haben gerade eine Datei in Galaxy importiert. Diese Datei ähnelt den Daten, die wir direkt von einer Sequenziereinrichtung erhalten könnten: eine FASTQ-Datei.
Praktische Übung: Inspect the FASTQ file
Überprüfen Sie die Datei, indem Sie auf das galaxy-eye (Auge) Symbol
Obwohl es kompliziert aussieht (und es vielleicht auch ist), ist das FASTQ-Format mit ein wenig Dekodierung leicht zu verstehen.
Jeder Read, der ein Fragment der Bibliothek darstellt, wird durch 4 Zeilen kodiert:
Line
Description
1
Always begins with @ followed by the information about the read
2
The actual nucleic sequence
3
Always begins with a + and contains sometimes the same info in line 1
4
Has a string of characters which represent the quality scores associated with each base of the nucleic sequence; must have the same number of characters as line 2
Die erste Sequenz in unserer Datei ist also zum Beispiel:
Das bedeutet, dass das Fragment mit der Bezeichnung @M00970 der DNA-Sequenz GTGCCAGCCGCCGCGGTAGTCCGACGTGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACCGAAGAACATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAAAAAACAAAAAAAAAAAAAGAAGCAAATGACGATTCAAGAAAGAAAAAAACACAGAATACTAACAATAAGTCATAAACATCATCAACATAAAAAAGGAAATACACTTACAACACATATCAATATCTAAAATAAATGATCAGCACACAACATGACGATTACCACACATGTGTACTACAAGTCAACTA entspricht und diese Sequenz mit einer Qualität von GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGFGGGGGGAFFGGFGGGGGGGGFGGGGGGGGGGGGGGFGGG+38+35*311*6,,31=******441+++0+0++0+*1*2++2++0*+*2*02*/***1*+++0+0++38++00++++++++++0+0+2++*+*+*+*+*****+0**+0**+***+)*.***1**//*)***)/)*)))*)))*),)0(((-((((-.(4(,,))).,(())))))).)))))))-))-( sequenziert wurde.
Aber was bedeutet dieser Qualitätswert?
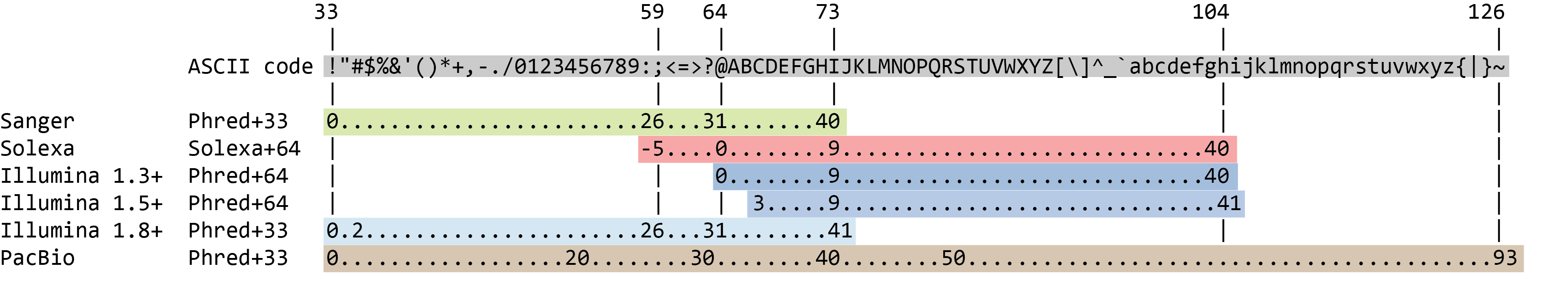
Der Qualitätsscore für jede Sequenz ist eine Zeichenkette, eine für jede Base der Nukleotidsequenz, um die Wahrscheinlichkeit einer falschen Identifizierung jeder Base zu charakterisieren. Der Score wird unter Verwendung der ASCII-Zeichentabelle (mit einigen historischen Unterschieden) kodiert:
Um Platz zu sparen, zeichnet der Sequenzer ein ASCII-Zeichen auf, um die Punktzahlen 0-42 darzustellen. Zum Beispiel entspricht 10 einem “+” und 40 einem “I”. FastQC weiß, wie dies zu übersetzen ist. Dies wird oft als “Phred”-Bewertung bezeichnet.
Jedem Nukleotid ist also ein ASCII-Zeichen zugeordnet, das seinen Phred-Qualitätsscore, die Wahrscheinlichkeit eines falschen Basenrufs, darstellt:
Phred Quality Score
Probability of incorrect base call
Base call accuracy
10
1 in 10
90%
20
1 in 100
99%
30
1 in 1000
99.9%
40
1 in 10,000
99.99%
50
1 in 100,000
99.999%
60
1 in 1,000,000
99.9999%
Was bedeutet 0-42? Wenn man diese Zahlen in eine Formel einsetzt, erhält man die Fehlerwahrscheinlichkeit für diese Basis. Die Formel lautet wie folgt: Q ist unser Qualitätswert (0-42) und P ist die Fehlerwahrscheinlichkeit:
Q = -10 log10(P)
Mit dieser Formel können wir berechnen, dass eine Qualitätsbewertung von 40 nur 0,00010 Wahrscheinlichkeit eines Fehlers bedeutet!
Frage
Welches ASCII-Zeichen entspricht dem schlechtesten Phred-Score für Illumina 1.8+?
Wie hoch ist der Phred-Qualitätsscore des 3. Nukleotids der ersten Sequenz?
Wie berechnet man die Genauigkeit der Nukleotidbase mit dem ASCII-Code /?
Wie hoch ist die Genauigkeit dieses 3. Nukleotids?
Der schlechteste Phred-Score ist der kleinste, also 0. Bei Illumina 1.8+ entspricht er dem Zeichen !.
Das 3. Nukleotid der 1. Sequenz hat ein ASCII-Zeichen G, was einem Score von 38 entspricht.
Dies kann wie folgt berechnet werden:
ASCII-Code für / ist 47
Qualitätsbewertung = 47-33=14
Formel zur Ermittlung der Fehlerwahrscheinlichkeit: \(P = 10^{-Q/10}\)
Das entsprechende Nukleotid G hat eine Genauigkeit von fast 96%
Kommentar
Das aktuelle lllumina (1.8+) verwendet das Sanger-Format (Phred+33). Wenn Sie mit älteren Datensätzen arbeiten, können Sie auf die älteren Scoring-Schemata stoßen. *mit *FastQC** tool, einem Tool, das wir später in diesem Tutorial verwenden werden, kann versucht werden, die Art der verwendeten Qualitätskodierung zu bestimmen (durch Bewertung des Bereichs der Phred-Werte in der FASTQ).
Wenn man sich die Datei in Galaxy ansieht, sieht es so aus, als ob die meisten Nukleotide einen hohen Score haben (G entspricht einem Score von 38). Gilt das für alle Sequenzen? Und entlang der gesamten Sequenzlänge?
Bewertung der Qualität mit FASTQE 🧬😎 - nur kurze Reads
Um einen Blick auf die Sequenzqualität entlang aller Sequenzen zu werfen, können wir FASTQE verwenden. Es ist ein Open-Source-Tool, das eine einfache und unterhaltsame Möglichkeit zur Qualitätskontrolle von Rohsequenzdaten bietet und diese als Emoji ausgibt. Sie können es verwenden, um einen schnellen Eindruck davon zu bekommen, ob Ihre Daten irgendwelche Probleme aufweisen, auf die Sie achten sollten, bevor Sie eine weitere Analyse durchführen.
Praktische Übung: Qualitätsprüfung
FASTQE ( Galaxy version 0.3.1+galaxy0) mit den folgenden Parametern
param-files“FastQ-Daten “: Reads
param-select“Anzuzeigende Score-Typen “: Mean
Prüfen Sie die generierte HTML-Datei
Anstatt die Qualitätswerte für jeden einzelnen Read zu betrachten, betrachtet FASTQE die Qualität kollektiv für alle Reads innerhalb einer Probe und kann den Mittelwert für jede Nukleotidposition entlang der Länge der Reads berechnen. Nachfolgend sind die Mittelwerte für diesen Datensatz aufgeführt.
Sie können den Score für jedes Emoji in der fastqe-Dokumentation sehen. Die folgenden Emojis mit Phred-Scores unter 20 sind diejenigen, von denen wir hoffen, dass wir sie nicht oft sehen werden.
Phred Quality Score
ASCII code
Emoji
0
!
🚫
1
”
❌
2
#
👺
3
$
💔
4
%
🙅
5
&
👾
6
’
👿
7
(
💀
8
)
👻
9
*
🙈
10
+
🙉
11
,
🙊
12
-
🐵
13
.
😿
14
/
😾
15
0
🙀
16
1
💣
17
2
🔥
18
3
😡
19
4
💩
Frage
Was ist der niedrigste Durchschnittswert in diesem Datensatz?
Der niedrigste Wert in diesem Datensatz ist 😿 13.
Bewertung der Qualität mit FastQC - kurze und lange Reads
Eine zusätzliche oder alternative Möglichkeit zur Überprüfung der Sequenzqualität ist FastQC. Es bietet eine modulare Reihe von Analysen, die Sie verwenden können, um zu prüfen, ob Ihre Daten Probleme aufweisen, auf die Sie achten sollten, bevor Sie eine weitere Analyse durchführen. Wir können damit zum Beispiel prüfen, ob bekannte Adapter in den Daten vorhanden sind. Wir führen es mit der FASTQ-Datei aus.
Praktische Übung: Qualitätsprüfung
FASTQC ( Galaxy version 0.73+galaxy0) mit den folgenden Parametern
param-files“Rohe Lesedaten aus Ihrer aktuellen Historie “: Reads
Prüfen Sie die generierte HTML-Datei
Frage
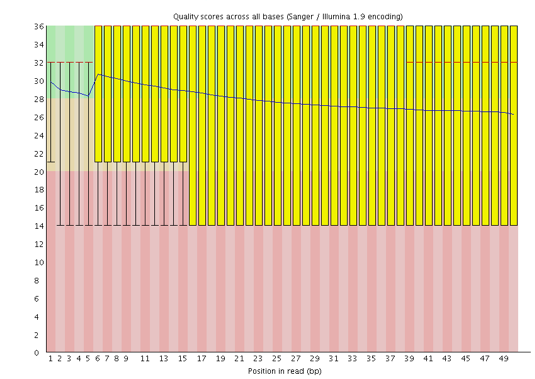
Welche Phred-Kodierung wird in der FASTQ-Datei für diese Sequenzen verwendet?
Die Phred-Scores werden mit Sanger / Illumina 1.9 (Encoding in der oberen Tabelle) kodiert.
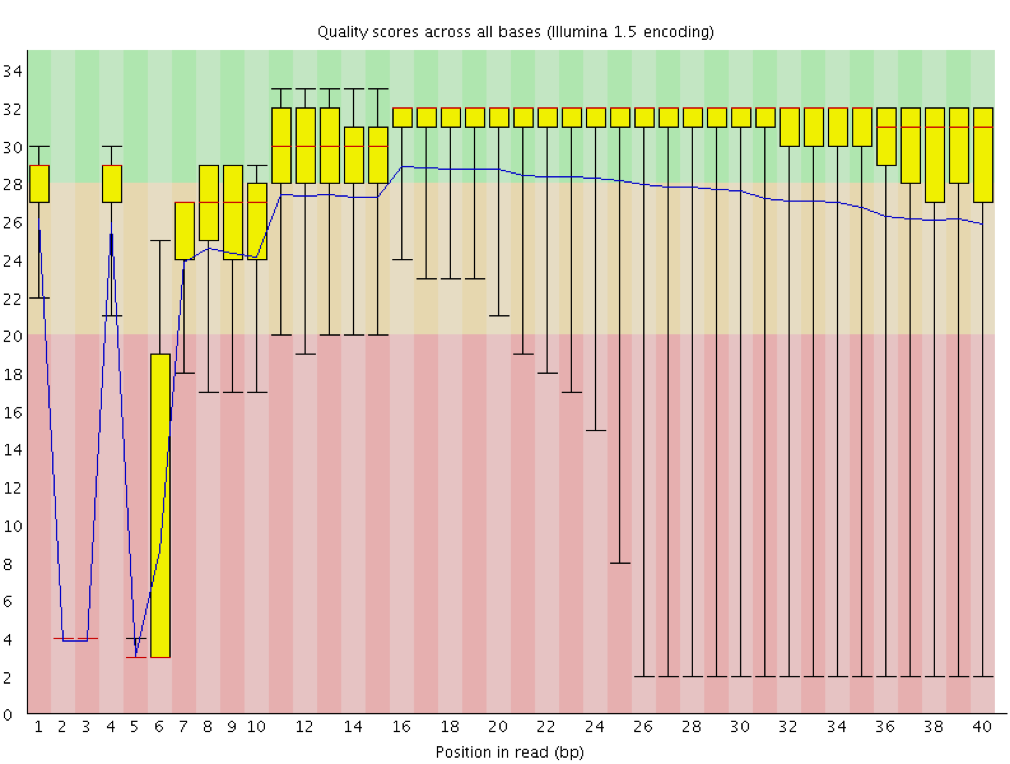
Qualität der Sequenz pro Base
Mit FastQC können wir die Basenqualität der Reads anhand des Plots für die Sequenzqualität pro Base überprüfen, ähnlich wie wir es mit FASTQE gemacht haben.
Auf der x-Achse ist die Basenposition im Read angegeben. In diesem Beispiel enthält die Probe Reads, die bis zu 296 bp lang sind.
Die x-Achse ist nicht immer gleichmäßig. Bei langen Leseabschnitten wird ein gewisses Binning angewendet, um die Dinge kompakt zu halten. Wir können das in unserer Probe sehen. Sie beginnt mit einzelnen 1-10 Basen. Danach werden die Basen über ein Fenster mit einer bestimmten Anzahl von Basen gebinnt. Datenbinning bedeutet Gruppierung und ist eine Technik der Datenvorverarbeitung, um die Auswirkungen kleinerer Beobachtungsfehler zu verringern. Die Anzahl der zusammengebinnten Basenpositionen hängt von der Länge des Reads ab. Bei Reads mit einer Länge von mehr als 50bp werden im letzten Teil des Diagramms aggregierte Statistiken für 5bp-Fenster angezeigt. Kürzere Reads haben kleinere Fenster und längere Reads größere Fenster. Das Binning kann bei der Ausführung von FastQC entfernt werden, indem der Parameter “Disable grouping of bases for reads >50bp” auf Yes gesetzt wird.
Für jede Position wird ein Boxplot mit gezeichnet:
der Medianwert, dargestellt durch die zentrale rote Linie
der Interquartilsbereich (25-75%), dargestellt durch den gelben Kasten
die 10%- und 90%-Werte in den oberen und unteren Whiskern
die mittlere Qualität, dargestellt durch die blaue Linie
Die y-Achse zeigt die Qualitätswerte an. Je höher die Punktzahl, desto besser ist der Base-Call. Der Hintergrund des Diagramms unterteilt die y-Achse in sehr gute Qualitätswerte (grün), Werte von angemessener Qualität (orange) und Werte von schlechter Qualität (rot).
Bei allen Illumina-Sequenzern ist es normal, dass der mittlere Qualitätswert in den ersten 5-7 Basen niedriger ist und dann ansteigt. Die Qualität der Reads nimmt bei den meisten Plattformen am Ende des Reads ab. Dies ist häufig auf einen Signalabfall oder eine Phasenverschiebung während des Sequenzierungslaufs zurückzuführen. Die jüngsten Entwicklungen in der Sequenzierungschemie haben dies etwas verbessert, aber die Reads sind jetzt länger als je zuvor.
Signalverfall
Die Intensität des Fluoreszenzsignals nimmt mit jedem Zyklus des Sequenziervorgangs ab. Aufgrund der sich abbauenden Fluorophore wird ein Teil der Stränge im Cluster nicht verlängert. Der Anteil des emittierten Signals nimmt mit jedem Zyklus weiter ab, was zu einer Verringerung der Qualitätswerte am 3’-Ende des Read führt.
Phasierung
Das Signal beginnt mit zunehmender Zykluszahl zu verschwimmen, weil der Cluster an Synchronität verliert. Im Laufe der Zyklen kommt es bei einigen Strängen zu zufälligen Ausfällen bei der Einbindung von Nukleotiden aufgrund von:
Unvollständige Entfernung der 3’-Terminatoren und Fluorophore
Einbau von Nukleotiden ohne wirksame 3’-Terminatoren
Dies führt zu einer Verringerung der Qualitätswerte am 3’-Ende des Reads.
Dies sind einige Qualitätsprofile pro Basensequenz, die auf Probleme mit der Sequenzierung hinweisen können.
Overclustering
Sequenziereinrichtungen können die Fließzellen überclustern. Dies führt zu geringen Abständen zwischen den Clustern und einer Überlappung der Signale. Zwei Cluster können als ein einziger Cluster interpretiert werden, wobei gemischte Fluoreszenzsignale erkannt werden und die Signalreinheit abnimmt. Dies führt zu niedrigeren Qualitätswerten für den gesamten Read.
Aufschlüsselung nach Instrumenten
Während eines Laufs kann es gelegentlich zu Problemen mit den Sequenziergeräten kommen. Ein plötzlicher Qualitätsabfall oder ein hoher Prozentsatz von Reads mit niedriger Qualität im gesamten Read könnte auf ein Problem in der Einrichtung hinweisen. Einige Beispiele für solche Probleme:
Manifold Burst
Zyklenverlust
Read 2 failure
Bei solchen Daten sollte die Sequenziereinrichtung kontaktiert werden, um sie zu besprechen. Oft ist dann eine erneute Sequenzierung erforderlich (und wird unserer Erfahrung nach auch von der Firma angeboten).
Frage
Wie verändert sich der mittlere Qualitätswert entlang der Sequenz?
Ist diese Tendenz bei allen Sequenzen zu beobachten?
Der mittlere Qualitätswert (blaue Linie) fällt etwa in der Mitte dieser Sequenzen ab. Es ist üblich, dass die mittlere Qualität zum Ende der Sequenzen hin abnimmt, da die Sequenzer am Ende mehr falsche Nukleotide einbauen. In dieser Probe gibt es jedoch einen sehr starken Qualitätsabfall ab der Mitte.
Die Box-Plots werden ab Position ~100 immer breiter. Das bedeutet, dass bei vielen Sequenzen die Punktzahl ab der Mitte der Sequenz abfällt. Nach 100 Nukleotiden haben mehr als 10% der Sequenzen Scores unter 20.
Wenn der Median der Qualität unter einem Phred-Score von ~20 liegt, sollten wir in Erwägung ziehen, Basen schlechter Qualität aus der Sequenz zu entfernen. Wir werden diesen Prozess im Abschnitt Trimmen und Filtern erläutern.
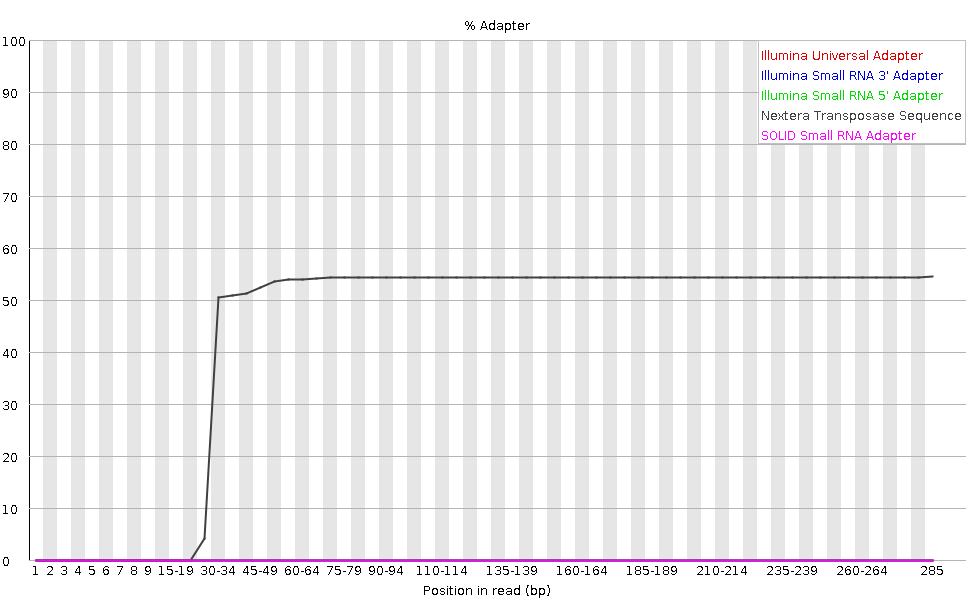
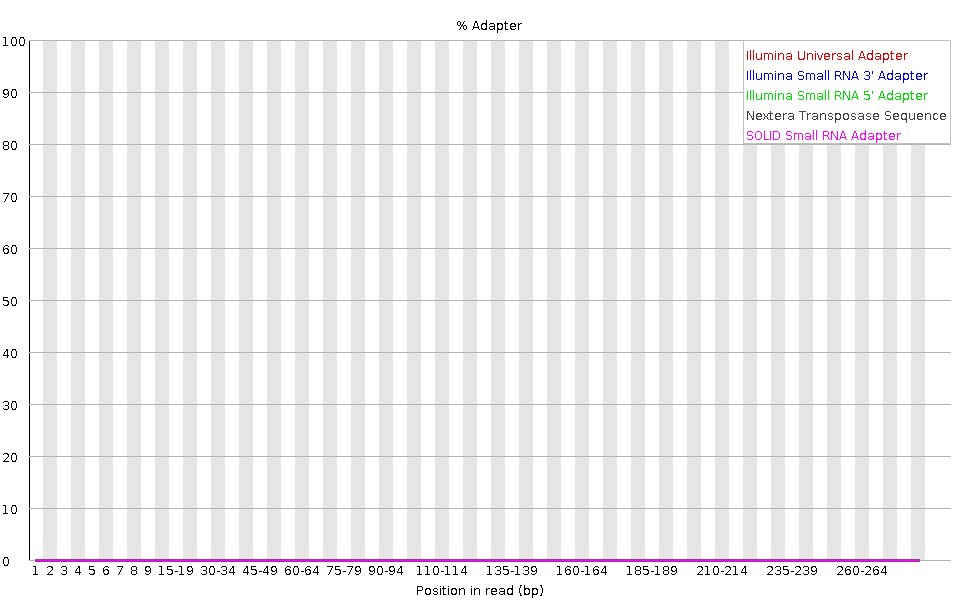
Die Grafik zeigt den kumulativen Prozentsatz der Reads mit den verschiedenen Adaptersequenzen an jeder Position. Sobald eine Adaptersequenz in einem Read vorkommt, wird sie bis zum Ende des Reads gezählt, so dass der Prozentsatz mit der Readlänge zunimmt. FastQC kann einige Adapter standardmäßig erkennen (z.B. Illumina, Nextera), für andere können wir eine Kontaminanten-Datei als Eingabe für das FastQC-Tool bereitstellen.
Idealerweise sollten Illumina-Sequenzdaten keine Adaptersequenz enthalten. Aber bei langen Reads sind einige der Bibliotheksinserts kürzer als die Leselänge, was zu einem Durchlesen des Adapters am 3’-Ende des Reads führt. Diese Mikrobiom-Probe hat relativ lange Reads und wir können sehen, dass der Nextera dapater erkannt wurde.
Der Adaptergehalt kann auch bei RNA-Seq-Bibliotheken festgestellt werden, bei denen die Verteilung der Bibliotheksinsertgrößen unterschiedlich ist und wahrscheinlich einige kurze Inserts enthält.
Wir können ein Trimming-Tool wie Cutadapt einsetzen, um diesen Adapter zu entfernen. Wir werden diesen Prozess im Abschnitt Filter und Trimmen erläutern.
In den folgenden Abschnitten werden einige der anderen von FastQC erzeugten Diagramme ausführlich beschrieben. Beachten Sie, dass einige Plots/Module Warnungen ausgeben können, aber für die Art der Daten, mit denen Sie arbeiten, normal sind, wie unten und in den FASTQC-FAQ beschrieben. Die anderen Diagramme geben uns Informationen, um die Qualität der Daten besser zu verstehen und um zu sehen, ob im Labor Änderungen vorgenommen werden können, um in Zukunft Daten von höherer Qualität zu erhalten. Diese Abschnitte sind optional, und wenn Sie sie überspringen möchten, können Sie das tun:
Springen Sie direkt zum nächsten Abschnitt, um mehr über das Trimmen von Paired-End-Daten zu erfahren
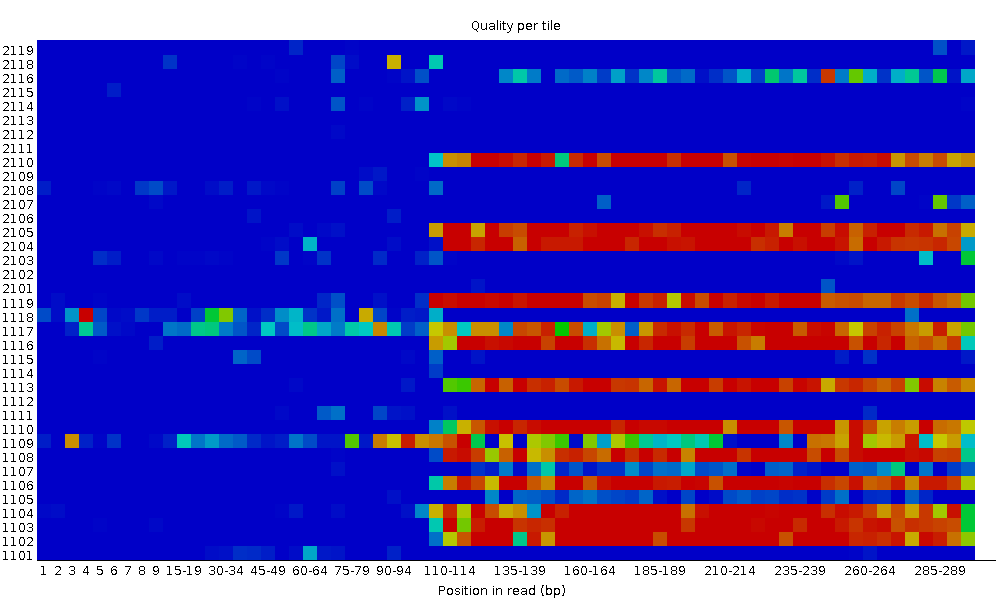
Qualität der Sequenz pro Kachel
Mit diesem Diagramm können Sie die Qualitätswerte der einzelnen Kacheln für alle Ihre Basen betrachten, um festzustellen, ob ein Qualitätsverlust nur mit einem Teil der Fließzelle verbunden ist. Das Diagramm zeigt die Abweichung von der durchschnittlichen Qualität für jede Fließzellenkachel an. Die wärmeren Farben zeigen an, dass die Reads in der jeweiligen Kachel eine schlechtere Qualität für diese Position aufweisen als die Reads in anderen Kacheln. Bei dieser Probe können Sie sehen, dass bestimmte Kacheln durchweg schlechte Qualität aufweisen, insbesondere ab ~100bp. Ein guter Plot sollte durchgängig blau sein.
Dieses Diagramm wird nur für Illumina-Bibliotheken angezeigt, die ihre ursprünglichen Sequenzkennungen beibehalten. Darin kodiert ist die Fließzellenkachel, aus der jeder Read stammt.
In einigen Fällen sind die bei der Sequenzierung verwendeten Chemikalien im Laufe der Zeit erschöpft und die letzten Kacheln haben die schlechtesten Chemikalien erhalten, was die Sequenzierungsreaktionen etwas fehleranfällig macht. Das Diagramm “Sequenzqualität pro Kachel” weist dann einige horizontale Linien wie diese auf:
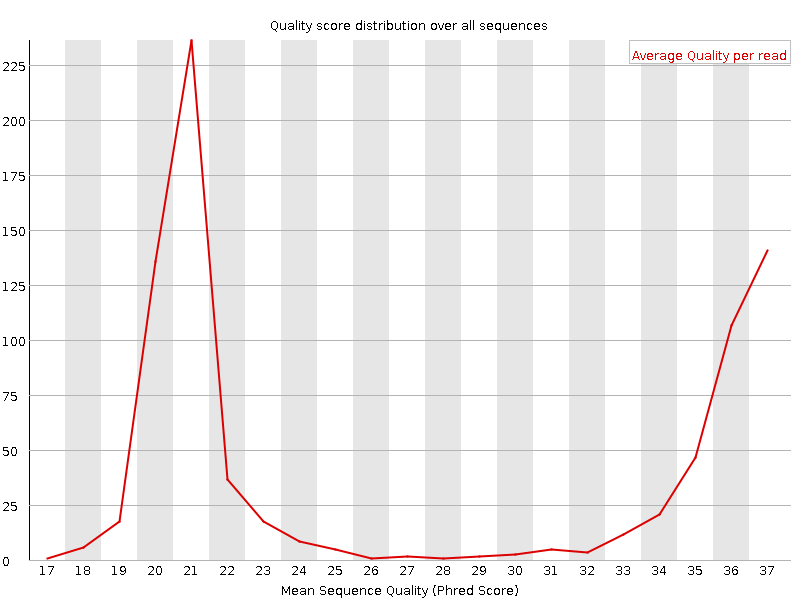
Qualitätswerte pro Sequenz
Auf der x-Achse ist die durchschnittliche Qualitätsbewertung über die gesamte Länge aller Reads aufgetragen, auf der y-Achse die Gesamtzahl der Reads mit dieser Bewertung:
Die Verteilung der durchschnittlichen Lesequalität sollte einen engen Peak im oberen Bereich des Diagramms aufweisen. Es kann auch angezeigt werden, wenn eine Teilmenge der Sequenzen durchgängig niedrige Qualitätswerte aufweist: Dies kann vorkommen, weil einige Sequenzen schlecht abgebildet werden (am Rande des Sichtfelds usw.), diese sollten jedoch nur einen kleinen Prozentsatz der gesamten Sequenzen ausmachen.
Abbildung 6: Inhalt pro Basensequenz für eine DNA-Bibliothek
“Per Base Sequence Content” stellt den prozentualen Anteil jedes der vier Nukleotide (T, C, A, G) an jeder Position über alle Reads in der Eingabesequenzdatei dar. Wie bei der Qualität der Sequenz pro Base ist die x-Achse uneinheitlich.
In einer zufälligen Bibliothek würde man erwarten, dass sich die vier Basen wenig bis gar nicht unterscheiden. Der Anteil jeder der vier Basen sollte über die Länge des Reads mit %A=%T und %G=%C relativ konstant bleiben, und die Linien in diesem Diagramm sollten parallel zueinander verlaufen. Es handelt sich um Amplikondaten, bei denen 16S-DNA mittels PCR amplifiziert und sequenziert wird. Daher ist zu erwarten, dass diese Darstellung eine gewisse Verzerrung aufweist und keine Zufallsverteilung zeigt.
Es ist erwähnenswert, dass einige Bibliothekstypen immer einn Sequenzzusammensetzungsbias erzeugen, normalerweise zu Beginn des Lesevorgangs. Bibliotheken, die durch Priming mit zufälligen Hexameren hergestellt wurden (einschließlich fast aller RNA-Seq-Bibliotheken), und solche, die mit Transposasen fragmentiert wurden, enthalten eine intrinsischen Bias an den Positionen, an denen Reads beginnen (die ersten 10-12 Basen). Dieser Bias bezieht sich nicht auf eine bestimmte Sequenz, sondern sorgt für eine Anreicherung einer Reihe verschiedener K-Mere am 5’-Ende der Reads. Dies ist zwar ein echter technischer Bias, kann aber nicht durch Trimming korrigiert werden und scheint in den meisten Fällen die nachgeschaltete Analyse nicht zu beeinträchtigen. Es wird jedoch eine Warnung oder ein Fehler in diesem Modul erzeugt.
Bei ChIP-seq-Daten kann es bei der Fragmentierung mit Transposasen auch zu Bias der Lesestartsequenz kommen. Bei Bisulfit-konvertierten Daten, z. B. HiC-Daten, wird eine Trennung von G von C und A von T erwartet:
Am Ende ist eine allgemeine Verschiebung in der Sequenzzusammensetzung festzustellen. Wenn die Verschiebung mit einem Verlust an Sequenzierqualität korreliert, kann vermutet werden, dass Fehlanrufe mit einem gleichmäßigerem Sequenzbias erfolgen als bei bisulfitkonvertierten Bibliotheken. Durch das Trimmen der Sequenzen wurde dieses Problem behoben, aber wenn dies nicht geschehen wäre, hätte es dramatische Auswirkungen auf die durchgeführten Methylierungs-Calls gehabt.
Frage
Warum gibt es eine Warnung bei den Graphen für den Sequenzgehalt pro Base?
Am Anfang der Sequenzen ist der Sequenzgehalt pro Base nicht wirklich gut und die Prozentsätze sind nicht gleich, wie für 16S-Amplikon-Daten erwartet.
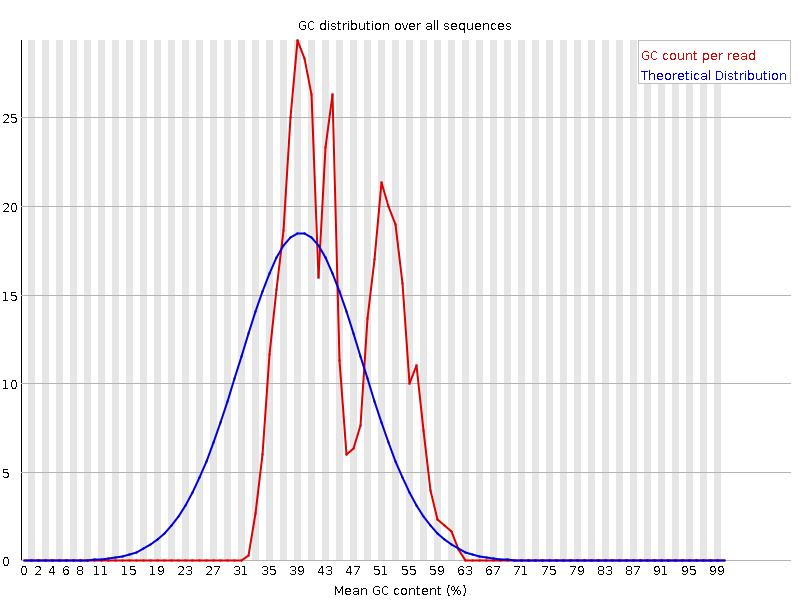
Diese Grafik zeigt die Anzahl der Reads im Vergleich zum Prozentsatz der Basen G und C pro Read. Sie wird mit einer theoretischen Verteilung verglichen, die von einem einheitlichen GC-Gehalt aller Reads ausgeht, wie er bei der Shotgun-Sequenzierung des gesamten Genoms erwartet wird, wobei der zentrale Peak dem gesamten GC-Gehalt des zugrunde liegenden Genoms entspricht. Da der GC-Gehalt des Genoms nicht bekannt ist, wird der modale GC-Gehalt aus den beobachteten Daten berechnet und zur Erstellung einer Referenzverteilung verwendet.
Eine ungewöhnlich geformte Verteilung könnte auf eine kontaminierte Bibliothek oder eine andere Art von Bias in der Teilmenge hinweisen. Eine verschobene Normalverteilung deutet auf einen systematischen Bias hin, der unabhängig von der Basenposition ist. Wenn es einen systematischen Bias gibt, der eine verschobene Normalverteilung erzeugt, wird dies vom Modul nicht als Fehler erkannt, da es nicht weiß, wie hoch der GC-Gehalt Ihres Genoms sein sollte.
Es gibt aber auch andere Situationen, in denen eine ungewöhnlich geformte Verteilung auftreten kann. Bei der RNA-Sequenzierung kann es beispielsweise zu einer mehr oder weniger starken Verteilung des mittleren GC-Gehalts unter den Transkripten kommen, was dazu führt, dass die beobachtete Kurve breiter oder schmaler ist als eine ideale Normalverteilung.
Frage
Warum sind die Graphen für den GC-Gehalt pro Sequenz fehlerhaft?
Es gibt mehrere Peaks. Dies kann auf unerwartete Kontaminationen wie Adapter, rRNA oder überrepräsentierte Sequenzen hinweisen. Es kann aber auch normal sein, wenn es sich um Amplikon-Daten handelt oder Sie sehr reichhaltige RNA-seq-Transkripte haben.
Sequenzlängenverteilung
Diese Grafik zeigt die Verteilung der Fragmentgrößen in der untersuchten Datei. In vielen Fällen ergibt sich ein einfaches Diagramm, das einen Peak nur bei einer Größe zeigt, aber bei FASTQ-Dateien mit variabler Länge zeigt es die relativen Mengen der einzelnen Sequenzfragmente unterschiedlicher Größe. Unser Diagramm zeigt die variable Länge, da wir die Daten getrimmt haben. Der größte Peak liegt bei 296bp, aber es gibt einen zweiten großen Peak bei ~100bp. Obwohl unsere Sequenzen bis zu 296bp lang sind, sind viele der qualitativ hochwertigen Sequenzen kürzer. Dies entspricht dem Abfall der Sequenzqualität bei ~100bp und den roten Streifen, die an dieser Position im Diagramm der Sequenzqualität pro Kachel beginnen.
Einige Hochdurchsatz-Sequenzer erzeugen Sequenzfragmente von einheitlicher Länge, andere können jedoch Reads von sehr unterschiedlicher Länge enthalten. Selbst innerhalb von Bibliotheken mit einheitlicher Länge schneiden einige Pipelines Sequenzen zu, um Basen-Calls von schlechter Qualität vom Ende oder den ersten \(n\) Basen zu entfernen, wenn sie mit den ersten \(n\) Basen des Adapters bis zu 90% übereinstimmen (standardmäßig), wobei manchmal \(n = 1\).
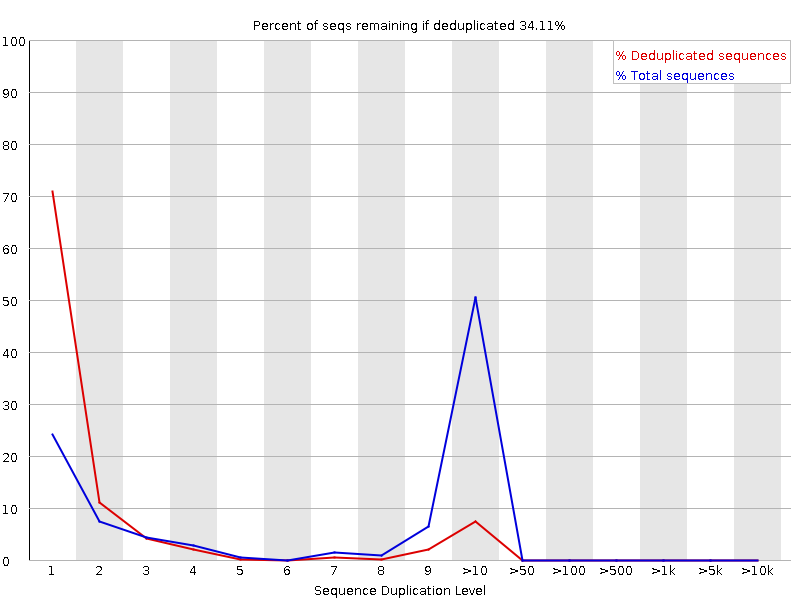
Sequenzduplikationsstufen
Das Diagramm zeigt in Blau den Prozentsatz der Reads einer bestimmten Sequenz in der Datei, die eine bestimmte Anzahl von Malen in der Datei vorhanden sind:
In einer heterogenen Bibliothek werden die meisten Sequenzen nur einmal im endgültigen Satz vorkommen. Eine geringe Duplizierung kann auf einen sehr hohen Abdeckungsgrad der Zielsequenz hinweisen, eine hohe Duplizierung deutet jedoch eher auf eine Art Anreicherungsbias hin.
Es lassen sich zwei Quellen für doppelte Reads finden:
PCR-Duplikation, bei der Bibliotheksfragmente aufgrund eines PCR-Anreicherungs-bias überrepräsentiert sind
Dies ist besorgniserregend, weil PCR-Duplikate den wahren Anteil der Sequenzen in der Eingabe falsch darstellen.
Wirklich überrepräsentierte Sequenzen, z. B. sehr häufig vorkommende Transkripte in einer RNA-Seq-Bibliothek oder in Amplikon-Daten (wie diese Probe)
Dies ist ein erwarteter Fall und nicht besorgniserregend, da er die Eingabe getreu darstellt.
FastQC zählt den Grad der Duplikation für jede Sequenz in einer Bibliothek und erstellt ein Diagramm, das die relative Anzahl der Sequenzen mit verschiedenen Duplikationsgraden anzeigt. Das Diagramm besteht aus zwei Linien:
Blaue Linie: Verteilung der Duplikationsgrade für den vollständigen Sequenzsatz
Rote Linie: Verteilung der entduplizierten Sequenzen mit den Anteilen der entduplizierten Menge, die aus verschiedenen Duplikationsstufen in den Originaldaten stammen.
Bei Shotgun-Daten für das gesamte Genom ist davon auszugehen, dass nahezu 100 % der Reads einzigartig sind (d. h. nur ein einziges Mal in den Sequenzdaten vorkommen). Die meisten Sequenzen sollten sowohl in der roten als auch in der blauen Linie ganz links im Diagramm liegen. Dies deutet auf eine hochdiverse Bibliothek hin, die nicht übersequenziert wurde. Wenn die Sequenzierungstiefe extrem hoch ist (z. B. > 100x die Größe des Genoms), kann es zu unvermeidlichen Sequenzduplikationen kommen: Theoretisch gibt es nur eine endliche Anzahl von völlig eindeutigen Sequenzlesungen, die aus einer gegebenen Eingangs-DNA-Probe gewonnen werden können.
Spezifischere Anreicherungen von Untergruppen oder das Vorhandensein von Verunreinigungen mit geringer Komplexität führen zu Spitzen auf der rechten Seite des Diagramms. Diese hohen Duplikationspeaks erscheinen meist in der blauen Kurve, da sie einen hohen Anteil der ursprünglichen Bibliothek ausmachen, verschwinden aber normalerweise in der roten Kurve, da sie einen unbedeutenden Anteil der deduplizierten Menge ausmachen. Bleiben die Peaks in der roten Kurve bestehen, deutet dies darauf hin, dass es eine große Anzahl verschiedener hoch duplizierter Sequenzen gibt, was entweder auf einen kontaminierten Satz oder eine sehr starke technische Duplikation hinweisen könnte.
Bei der RNA-Sequenzierung gibt es in der Regel einige sehr häufig vorkommende Transkripte und einige weniger häufig vorkommende. Es ist zu erwarten, dass doppelte Reads für Transkripte mit hoher Häufigkeit beobachtet werden:
Überrepräsentierte Sequenzen
Eine normale Bibliothek mit hohem Durchsatz enthält eine Vielzahl von Sequenzen, wobei keine einzelne Sequenz einen winzigen Bruchteil des Ganzen ausmacht. Die Feststellung, dass eine einzelne Sequenz in der Menge stark überrepräsentiert ist, bedeutet entweder, dass sie biologisch sehr bedeutsam ist, oder sie weist darauf hin, dass die Bibliothek kontaminiert oder nicht so vielfältig ist wie erwartet.
FastQC listet alle Sequenzen auf, die mehr als 0,1 % der Gesamtmenge ausmachen. Für jede überrepräsentierte Sequenz sucht FastQC nach Übereinstimmungen in einer Datenbank mit üblichen Verunreinigungen und meldet den besten gefundenen Treffer. Die Treffer müssen mindestens 20bp lang sein und dürfen nicht mehr als 1 Mismatch aufweisen. Das Auffinden eines Treffers bedeutet nicht unbedingt, dass es sich um die Quelle der Verunreinigung handelt, aber es kann Ihnen einen Hinweis auf die richtige Richtung geben. Es ist auch erwähnenswert, dass viele Adaptersequenzen einander sehr ähnlich sind, so dass ein Treffer gemeldet werden kann, der technisch nicht korrekt ist, aber eine sehr ähnliche Sequenz wie die eigentliche Übereinstimmung aufweist.
RNA-Sequenzierungsdaten können einige Transkripte enthalten, die so häufig vorkommen, dass sie als überrepräsentierte Sequenz registriert werden. Bei DNA-Sequenzierungsdaten sollte keine einzelne Sequenz so häufig vorkommen, dass sie aufgelistet wird, aber wir können manchmal einen kleinen Prozentsatz von Adapter-Reads sehen.
Frage
Wie können wir herausfinden, welche Sequenzen überrepräsentiert sind?
Wir können überrepräsentierte Sequenzen mit BLAST untersuchen, um zu sehen, um welche es sich handelt. In diesem Fall nehmen wir die am stärksten überrepräsentierte Sequenz
und blastn gegen die Standard-Nukleotiddatenbank (nr/nt) verwenden, erhalten wir keine Treffer. Aber wenn wir VecScreen verwenden, sehen wir, dass es der Nextera-Adapter ist.
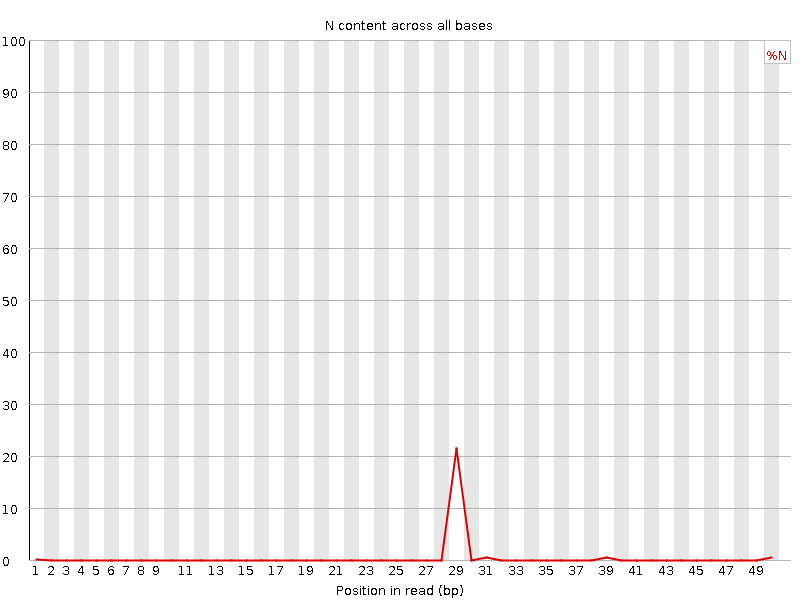
Wenn ein Sequenzer nicht in der Lage ist, einen Base-Call mit ausreichender Sicherheit durchzuführen, schreibt er ein “N” anstelle eines herkömmlichen Base-Calls. Dieses Diagramm zeigt den Prozentsatz der Basenaufrufe an jeder Position oder jedem Bin an, für die ein “N” aufgerufen wurde.
Es ist nicht ungewöhnlich, dass ein sehr hoher Anteil von Ns in einer Sequenz auftritt, insbesondere gegen Ende der Sequenz. Aber diese Kurve sollte niemals merklich über Null ansteigen. Wenn dies der Fall ist, deutet dies auf ein Problem während des Sequenzierungslaufs hin. Im folgenden Beispiel führte ein Fehler dazu, dass das Gerät bei etwa 20 % der Reads an Position 29 keine Base aufrufen konnte:
Kmer-Gehalt
Dieser Plot wird standardmäßig nicht ausgegeben. Wie im Tool-Formular angegeben, muss dieses Modul mit Hilfe eines benutzerdefinierten Submoduls und einer Grenzwertdatei aktiviert werden, wenn Sie es wünschen. Mit diesem Modul führt FastQC eine generische Analyse aller kurzen Nukleotidsequenzen der Länge k (kmer, standardmäßig k = 7) durch, beginnend an jeder Position entlang des Reads in der Bibliothek, um diejenigen zu finden, die keine gleichmäßige Abdeckung über die Länge Ihrer Reads haben. Jedes gegebene kmer sollte gleichmäßig über die Länge des Reads vertreten sein.
FastQC meldet die Liste der kmers, die an bestimmten Positionen mit einer größeren Häufigkeit als erwartet auftreten. Dies kann auf verschiedene Quellen von Bias in der Bibliothek zurückzuführen sein, einschließlich des Vorhandenseins von Readthrough-Adaptersequenzen, die sich am Ende der Sequenzen ansammeln. Das Vorhandensein von überrepräsentierten Sequenzen in der Bibliothek (z. B. Adapterdimere) führt dazu, dass das Kmer-Diagramm von den Kmer-Werten dieser Sequenzen dominiert wird. Kmere, die aufgrund anderer interessanter Biases verzerrt sind, können dann verwässert werden und sind nicht leicht zu erkennen.
Das folgende Beispiel stammt aus einer hochwertigen DNA-Seq-Bibliothek. Die biased kmers in der Nähe des Lesebeginns sind wahrscheinlich auf eine geringe sequenzabhängige Effizienz der DNA-Scherung oder auf ein zufälliges Priming zurückzuführen:
Dieses Modul kann sehr schwer zu interpretieren sein. Der Adapter-Content-Plot und die Tabelle der überrepräsentierten Sequenzen sind einfacher zu interpretieren und können Ihnen genügend Informationen liefern, ohne dass Sie diesen Plot benötigen. RNA-seq-Bibliotheken können stark vertretene kmers haben, die von hoch exprimierten Sequenzen stammen. Um mehr über diese Darstellung zu erfahren, lesen Sie bitte die FastQC Kmer Content documentation.
Wir haben versucht, hier verschiedene FastQC-Berichte und einige Anwendungsfälle zu erklären. Mehr darüber und auch einige häufige Probleme bei Next-Generation-Sequenzierung finden Sie auf QCFAIL.com
Kleine/Mikro-RNA
In kleinen RNA-Bibliotheken haben wir in der Regel einen relativ kleinen Satz einzigartiger, kurzer Sequenzen. Kleine RNA-Bibliotheken werden vor dem Hinzufügen von Sequenzieradaptern an ihren Enden nicht zufällig geschert: Alle Reads für bestimmte Klassen von microRNAs werden identisch sein. Das Ergebnis wird sein:
Extrem verzerrter Inhalt pro Basensequenz
Äußerst enge Verteilung des GC-Gehalts
Sehr hohe Sequenzduplikationsraten
Häufigkeit der überrepräsentierten Sequenzen
Durchlesen in Adaptern
Amplikon
Amplikon-Bibliotheken werden durch PCR-Amplifikation eines bestimmten Ziels hergestellt. Zum Beispiel die hypervariable Region V4 des bakteriellen 16S rRNA-Gens. Es wird erwartet, dass alle Reads aus dieser Art von Bibliothek nahezu identisch sind. Das Ergebnis wird sein:
Extrem verzerrter Inhalt pro Basensequenz
Äußerst enge Verteilung des GC-Gehalts
Sehr hohe Sequenzduplikationsraten
Häufigkeit der überrepräsentierten Sequenzen
Bisulfit- oder Methylierungssequenzierung
Bei der Bisulfit- oder Methylierungssequenzierung wird die Mehrzahl der Cytosinbasen (C) in Thyminbasen (T) umgewandelt. Das Ergebnis ist:
Verzerrter Inhalt pro Basensequenz
Verzerrter GC-Gehalt pro Sequenz
Adapter-Dimer-Kontamination
Jeder Bibliothekstyp kann einen sehr geringen Prozentsatz von Adapter-Dimer-Fragmenten (d. h. ohne Insert) enthalten. Sie sind eher in Amplikon-Bibliotheken zu finden, die vollständig durch PCR (durch Bildung von PCR-Primer-Dimern) aufgebaut sind, als in DNA-Seq- oder RNA-Seq-Bibliotheken, die durch Adapter-Ligation aufgebaut sind. Wenn ein ausreichender Anteil der Bibliothek aus Adapter-Dimeren besteht, wird dies im FastQC-Bericht sichtbar:
Abfall der Sequenzqualität pro Base nach Base 60
Mögliche bimodale Verteilung der Qualitätswerte pro Sequenz
Ausgeprägtes Muster im Sequenzgehalt pro Base bis zur Base 60
Spike im GC-Gehalt pro Sequenz
Überrepräsentierte Sequenz mit passendem Adapter
Adaptergehalt > 0% ab Basis 1
Kommentar: Schlechte Qualität der Sequenzen
Wenn die Qualität der Reads nicht gut ist, sollten wir immer zuerst prüfen, was falsch ist und darüber nachdenken: Es kann an der Art der Sequenzierung liegen oder an dem, was wir sequenziert haben (hohe Anzahl überrepräsentierter Sequenzen in Transkriptomikdaten, verzerrter Prozentsatz von Basen in HiC-Daten).
Sie können sich auch an die Sequenziereinrichtung wenden, insbesondere wenn die Qualität wirklich schlecht ist: Die Qualitätsbehandlungen können nicht alles lösen. Wenn zu viele Basen von schlechter Qualität weggeschnitten werden, werden die entsprechenden Reads herausgefiltert und Sie verlieren sie.
Trimmen und Filtern - kurze Reads
Die Qualität nimmt in der Mitte dieser Sequenzen ab. Dies könnte bei nachgelagerten Analysen zu einem Bias durch diese möglicherweise falsch benannten Nukleotide führen. Die Sequenzen müssen behandelt werden, um eine Verzerrung in der nachgelagerten Analyse zu vermeiden. Trimming kann dazu beitragen, die Anzahl der Reads zu erhöhen, die der Aligner oder Assembler erfolgreich nutzen kann, und die Anzahl der nicht gemappten oder nicht assemblierten Reads zu verringern. Im Allgemeinen umfassen die Qualitätsbehandlungen:
Trimmen/Schneiden/Maskieren von Sequenzen
aus Regionen mit niedriger Qualitätsbewertung
Anfang/Ende der Sequenz
Entfernen von Adaptern
Filterung von Sequenzen
mit niedriger mittlerer Qualitätsbewertung
zu kurz
mit zu vielen mehrdeutigen (N) Basen
Um diese Aufgabe zu bewältigen, werden wir CutadaptMarcel 2011 verwenden, ein Tool, das die Sequenzqualität durch automatisiertes Adapter-Trimming und Qualitätskontrolle verbessert. Wir werden:
Trimmen von Basen geringer Qualität an den Enden. Das Qualitätstrimming wird vor dem Adaptertrimming durchgeführt. Wir setzen den Qualitätsschwellenwert auf 20, ein häufig verwendeter Schwellenwert, siehe mehr in GATK’s Phred Score FAQ.
Trimmen des Adapters mit Cutadapt. Dazu müssen wir die Sequenz des Adapters angeben. In diesem Beispiel ist Nextera der Adapter, der erkannt wurde. Wir können die Sequenz des Nextera-Adapters auf der Illumina-Website hierCTGTCTCTTATACACATCT finden. Wir werden diese Sequenz vom 3’-Ende der Reads abschneiden.
Herausfiltern von Sequenzen mit einer Länge < 20 nach dem Trimmen
Praktische Übung: Verbesserung der Sequenzqualität
Cutadapt ( Galaxy version 4.9+galaxy1) mit den folgenden Parametern
Wenn Ihre FASTQ-Datei nicht ausgewählt werden kann, können Sie überprüfen, ob das Format FASTQ mit Sanger-skalierten Qualitätswerten (fastqsanger.gz) ist. Sie können den Datentyp bearbeiten, indem Sie auf das Bleistiftsymbol klicken.
param-select“Zusätzliche zu erzeugende Ausgaben “: Report
Prüfen Sie die erzeugte txt-Datei (Report)
Frage
Wie viel % der Reads enthalten Adapter?
Wie viel % der Reads wurden wegen schlechter Qualität abgeschnitten?
Wie viel % der Reads wurden entfernt, weil sie zu kurz waren?
56,8% der Reads enthalten Adapter (Reads with adapters:)
35,1% der Reads wurden wegen schlechter Qualität abgeschnitten (Quality-trimmed:)
0 % der Reads wurden entfernt, weil sie zu kurz waren
Einer der größten Vorteile von Cutadapt im Vergleich zu anderen Trimming-Tools (z.B. TrimGalore!) ist, dass es eine gute Dokumentation gibt, die die Funktionsweise des Tools im Detail erklärt.
Der Cutadapt-Algorithmus zum Trimmen der Qualität besteht aus drei einfachen Schritten:
Subtrahieren Sie den gewählten Schwellenwert vom Qualitätswert jeder Position
Berechnen Sie eine Teilsumme dieser Unterschiede vom Ende der Sequenz bis zu jeder Position (solange die Teilsumme negativ ist)
Schnitt beim Minimalwert der Partialsumme
Im folgenden Beispiel gehen wir davon aus, dass das 3’-Ende mit einem Schwellenwert von 10 qualitätsgekürzt werden soll und wir folgende Qualitätswerte haben
42 40 26 27 8 7 11 4 2 3
Subtrahieren Sie den Schwellenwert
32 30 16 17 -2 -3 1 -6 -8 -7
Addieren Sie die Zahlen, beginnend am 3’-Ende (Teilsummen) und hören Sie frühzeitig auf, wenn die Summe größer als Null ist
(70) (38) 8 -8 -25 -23 -20, -21 -15 -7
Die Zahlen in Klammern sind nicht berechnet (weil 8 größer als Null ist), werden hier aber der Vollständigkeit halber gezeigt.
Wählen Sie die Position des Minimums (-25) als Trimmposition
Daher wird der Read auf die ersten vier Basen beschnitten, die Qualitätswerte aufweisen
42 40 26 27
Man beachte, dass daher auch Positionen mit einem Qualitätswert größer als der gewählte Schwellenwert entfernt werden, wenn sie in Regionen mit geringerer Qualität eingebettet sind (die Teilsumme ist abnehmend, wenn die Qualitätswerte kleiner als der Schwellenwert sind). Der Vorteil dieses Verfahrens ist, dass es robust gegenüber einer kleinen Anzahl von Positionen mit einer Qualität über dem Schwellenwert ist.
Alternativen zu diesem Verfahren wären:
Schnitt nach der ersten Position mit einer Qualität kleiner als der Schwellenwert
Schiebefenster-Ansatz
Der Ansatz des gleitenden Fensters prüft, ob die durchschnittliche Qualität jedes Sequenzfensters einer bestimmten Länge größer als der Schwellenwert ist. Beachten Sie, dass dieser Ansatz im Gegensatz zu cutadapt einen weiteren Parameter hat und die Robustheit von der Länge des Fensters (in Kombination mit dem Qualitätsschwellenwert) abhängt. Beide Ansätze sind in Trimmomatic implementiert.
Wir können unsere beschnittenen Daten mit FASTQE und/oder FastQC untersuchen.
Praktische Übung: Überprüfung der Qualität nach dem Trimmen
FASTQE ( Galaxy version 0.3.1+galaxy0): Führen Sie FASTQE mit den folgenden Parametern erneut aus
param-files“FastQ-Daten “: Cutadapt Read 1 Output
param-select“Anzuzeigende Score-Typen “: Mean
Prüfen Sie den neuen FASTQE-Bericht
Frage
Vergleichen Sie die FASTQE-Ausgabe mit der vorherigen Ausgabe vor dem obigen Trimmen. Wurde die Sequenzqualität verbessert?
Wenn Sie zwei oder mehr Datensätze gleichzeitig betrachten möchten, können Sie die Funktion Fenster-Manager in Galaxy verwenden:
Klicken Sie auf das Symbol Fenstermanagergalaxy-scratchbook in der oberen Menüleiste.
Sie sollten jetzt ein kleines Häkchen auf dem Symbol sehen
Betrachtengalaxy-eye Sie einen Datensatz, indem Sie auf das Augensymbol galaxy-eye klicken, um die Ausgabe zu betrachten
Sie sollten die Ausgabe in einem Fenster sehen, das sich über Galaxy legt
Sie können die Größe dieses Fensters ändern, indem Sie an der rechten unteren Ecke ziehen
Klick außerhalb der Datei, um den Fenstermanager zu beenden
Betrachtengalaxy-eye einen zweiten Datensatz aus Ihrer Historie
Sie sollten nun ein zweites Fenster mit dem neuen Datensatz sehen
Dies erleichtert den Vergleich der beiden Ausgaben
Wiederholen Sie diesen Vorgang für so viele Dateien, wie Sie vergleichen möchten
Sie können den Fenstermanagergalaxy-scratchbook ausschalten, indem Sie erneut auf das Symbol klicken
Ja, die Emojis für die Qualitätsbewertung sehen jetzt besser (fröhlicher) aus.
Mit FastQC konnten wir die Qualität der Basen im Datensatz verbessern und den Adapter entfernen.
Wir haben einige rote Streifen, da wir diese Regionen aus den Reads herausgeschnitten haben.
Wir haben jetzt einen Peak hoher Qualität anstelle eines hohen und eines niedrigeren, wie wir ihn vorher hatten.
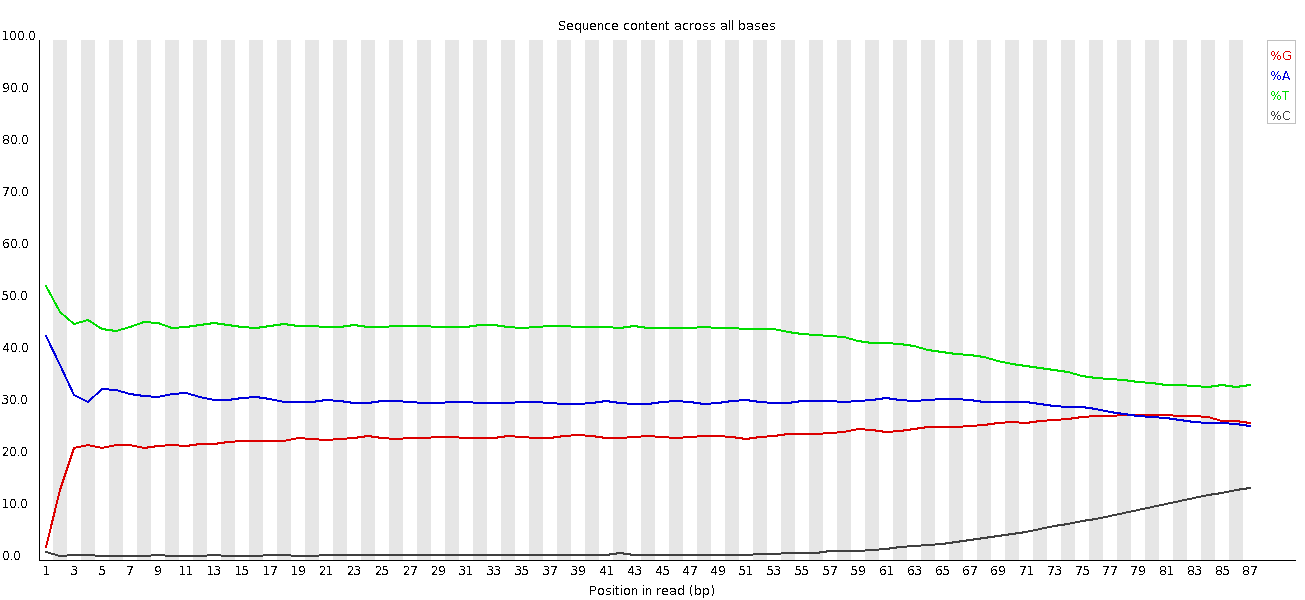
Da es sich um Amplikon-Daten handelt, sind die Basen nicht mehr gleichmäßig vertreten.
Wir haben jetzt einen einzigen Haupt-GC-Peak aufgrund der Entfernung des Adapters.
Dies ist dasselbe wie vorher, da wir keine Ns in diesen Reads haben.
Wir haben jetzt mehrere Peaks und eine Reihe von Längen, anstelle des einzelnen Peaks, den wir vor dem Trimmen hatten, als alle Sequenzen die gleiche Länge hatten.
Frage
Was entspricht der am stärksten überrepräsentierten Sequenz GTGTCAGCCGCCGCGGTAGTCCGACGTGG?
Wenn wir die am stärksten überrepräsentierte Sequenz nehmen
>overrep_seq1_after
GTGTCAGCCGCCGCGGTAGTCCGACGTGG
und blastn gegen die Standard-Nukleotiddatenbank (nr/nt) verwenden, sehen wir, dass die besten Treffer bei 16S rRNA-Genen zu finden sind. Dies ist sinnvoll, da es sich um 16S-Amplikon-Daten handelt, bei denen das 16S-Gen durch PCR amplifiziert wird.
Verarbeitung mehrerer Datensätze
Verarbeitung von Paired-End-Daten
Bei der Paired-End-Sequenzierung werden die Fragmente von beiden Seiten sequenziert. Dieser Ansatz führt zu zwei Reads pro Fragment, wobei der erste Read in Vorwärtsorientierung und der zweite Read in Rückwärtskomplement-Orientierung erfolgt. Mit dieser Technik haben wir den Vorteil, dass wir mehr Informationen über jedes DNA-Fragment erhalten, als wenn wir nur mit Single-End-Sequenzierung sequenzieren:
Der Abstand zwischen den beiden Reads ist bekannt und stellt daher eine zusätzliche Information dar, die die Zuordnung der Reads verbessern kann.
Die Paired-End-Sequenzierung erzeugt 2 FASTQ-Dateien:
Eine Datei mit den Sequenzen, die der vorwärts Orientierung aller Fragmente entsprechen
Eine Datei mit den Sequenzen, die der umgekehrten Orientierung aller Fragmente entsprechen
Normalerweise erkennen wir diese beiden Dateien, die zu einer Probe gehören, an dem Namen, der den gleichen Bezeichner für die Reads, aber eine unterschiedliche Erweiterung hat, z. B. sampleA_R1.fastq für die Forward Reads und sampleA_R2.fastq für die Reverse Reads. Es kann auch _f oder _1 für die Forward Reads und _r oder _2 für die Reverse Reads sein.
Die Daten, die wir im vorigen Schritt analysiert haben, waren Single-End-Daten, so dass wir einen Paired-End-RNA-seq-Datensatz importieren werden. Wir werden FastQC ausführen und die beiden Berichte mit MultiQC Ewels et al. 2016 aggregieren.
Praktische Übung: Bewertung der Qualität von Paired-End-Reads
Importieren Sie die Paired-End-Reads GSM461178_untreat_paired_subset_1.fastq und GSM461178_untreat_paired_subset_2.fastq von Zenodo oder aus der Datenbibliothek (fragen Sie Ihren Lehrer)
FASTQC ( Galaxy version 0.73+galaxy0) mit beiden Datensätzen:
param-files“Raw read data from your current history “: die beiden hochgeladenen Datensätze.
Klicken Sie auf param-filesMultiple datasets
Wählen Sie mehrere Dateien aus, indem Sie die Strg (oder COMMAND) Taste gedrückt halten und die gewünschten Dateien anklicken
MultiQC ( Galaxy version 1.9+galaxy1) mit den folgenden Parametern, um die FastQC-Berichte von Vorwärts- und Rückwärts-Reads zu aggregieren
In “Ergebnisse “
“Mit welchem Tool wurden die Protokolle erstellt? “: FastQC
In “FastQC output “
“Typ der FastQC-Ausgabe? “: Raw data
param-files“FastQC-Ausgabe “: Raw data-Dateien (Ausgabe der beiden FastQCtool)
Prüfen Sie die Webpage-Ausgabe von MultiQC.
Frage
Was halten Sie von der Qualität der Sequenzen?
Was sollten wir tun?
Die Qualität der Sequenzen scheint bei den Reverse-Reads schlechter zu sein als bei den Forward-Reads:
Per Sequence Quality Scores: Verteilung eher links, d. h. eine niedrigere mittlere Qualität der Sequenzen
Qualität der Sequenz pro Base: weniger glatte Kurve und stärkerer Abfall am Ende mit einem Mittelwert unter 28
Per Base Sequence Content: stärkere Verzerrung am Anfang und keine klare Unterscheidung zwischen C-G und A-T Gruppen
Die anderen Indikatoren (Adapter, Duplikationsgrad, etc.) sind ähnlich.
Wir sollten das Ende der Sequenzen abschneiden und sie mit Cutadapttool filtern
Bei Paired-End-Reads sind die durchschnittlichen Qualitätswerte für Forward-Reads fast immer höher als für Reverse-Reads.
Nach dem Trimmen sind die Reverse-Reads aufgrund ihrer Qualität kürzer und werden während des Filterungsschritts eliminiert. Wenn einer der Reverse-Reads entfernt wird, sollte auch der entsprechende Forward-Read entfernt werden. Andernfalls erhalten wir eine unterschiedliche Anzahl von Reads in beiden Dateien und in unterschiedlicher Reihenfolge, und die Reihenfolge ist für die nächsten Schritte wichtig. Daher ist es wichtig, die Forward- und Reverse-Reads beim Trimmen und Filtern gemeinsam zu behandeln.
Praktische Übung: Verbesserung der Qualität von Paired-End-Daten
Cutadapt ( Galaxy version 4.9+galaxy1) mit den folgenden Parametern
In diesen Datensätzen wurden keine Adapter gefunden. Wenn Sie Ihre eigenen Daten verarbeiten und wissen, welche Adaptersequenzen bei der Bibliotheksvorbereitung verwendet wurden, sollten Sie die Sequenzen hier angeben.
In “Other Read Trimming Options “
“Quality cutoff(s) (R1) “: 20
In “Read Filtering Options “
“Mindestlänge (R1) “: 20
param-select“Zusätzliche zu erzeugende Ausgaben “: Report
Prüfen Sie die erzeugte txt-Datei (Report)
Frage
Wie viele Basenpaare wurden aufgrund schlechter Qualität aus den Reads entfernt?
Wie viele Sequenzpaare wurden entfernt, weil sie zu kurz waren?
44.164 bp (Quality-trimmed:) für die Forward Reads und 138.638 bp für die Reverse Reads.
1.376 Sequenzen wurden entfernt, weil mindestens ein Read kürzer als der Längen-Cutoff war (322, wenn nur die Forward Reads analysiert wurden).
Zusätzlich zu dem Bericht erzeugt Cutadapt 2 Dateien:
Read 1 mit den getrimmten und gefilterten Forward Reads
Read 2 mit den getrimmten und gefilterten Reverse Reads
Diese Datensätze können für die nachgelagerte Analyse, z. B. für das Mapping, verwendet werden.
Frage
Welche Art von Alignment wird für die Suche nach Adaptern in Reads verwendet?
Welches ist das Kriterium für die Auswahl des besten Adapter-Alignments?
Semiglobales Alignment, d. h. nur der sich überlappende Teil der Lesung und der Adaptersequenz wird für die Auswertung verwendet.
Es wird ein Alignment mit maximaler Überlappung berechnet, das die geringste Anzahl von Mismatches und Indels aufweist.
Bewertung der Qualität mit Nanoplot - nur lange Reads
Bei langen Reads können wir die Sequenzqualität mit Nanoplot (De Coster et al. 2018) überprüfen. Es bietet grundlegende Statistiken mit schönen Diagrammen für einen schnellen Überblick über die Qualitätskontrolle.
Praktische Übung: Qualitätsprüfung von langen Reads
Erstellen Sie einen neuen Verlauf für diesen Teil und geben Sie ihm einen passenden Namen
Importieren Sie die PacBio HiFi-Reads m64011_190830_220126.Q20.subsample.fastq.gz von Zenodo
Abbildung 16: Comparison of Qscore between Illumina, PacBio and Nanopore
Definition: Qscores ist die durchschnittliche Fehlerwahrscheinlichkeit pro Base, ausgedrückt auf der log (Phred)-Skala
Was ist der Median, Mittelwert und N50?
Der Median, die mittlere Leselänge und auch N50 liegen nahe bei 18.000bp.
Bei PacBio HiFi-Reads liegt die Mehrheit der Reads im Allgemeinen in der Nähe dieses Wertes, da die Bibliotheksvorbereitung einen Schritt zur Größenauswahl umfasst. Bei anderen Technologien wie PacBio CLR und Nanopore ist der Wert größer und hängt hauptsächlich von der Qualität Ihrer DNA-Extraktion ab.
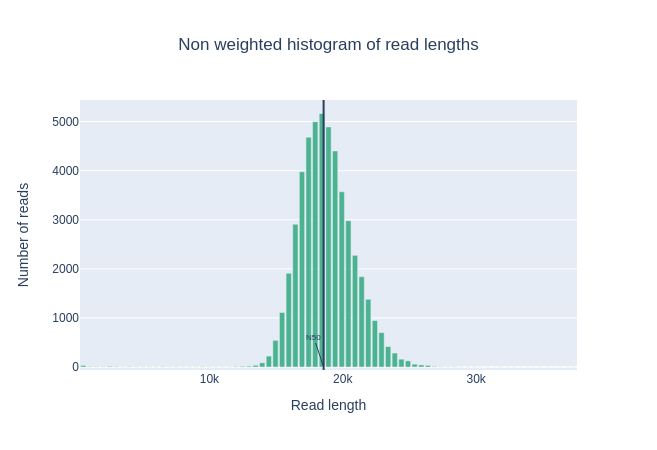
Histogramm der Leselängen
Diese Grafik zeigt die Verteilung der Fragmentgrößen in der analysierten Datei. Im Gegensatz zu den meisten Illumina-Läufen haben lange Reads eine variable Länge, und dies zeigt die relativen Mengen der einzelnen Sequenzfragmente unterschiedlicher Größe. In diesem Beispiel ist die Verteilung der Leselänge in der Nähe von 18kbp zentriert, aber die Ergebnisse können je nach Experiment sehr unterschiedlich sein.
Darstellung der Leselängen im Vergleich zur durchschnittlichen Lesequalität mit Punkten
Diese Darstellung zeigt die Verteilung der Fragmentgrößen entsprechend dem Qscore in der analysierten Datei. Im Allgemeinen gibt es keinen Zusammenhang zwischen Leselänge und Lesequalität, aber diese Darstellung ermöglicht es, beide Informationen in einem einzigen Diagramm zu visualisieren und mögliche Abweichungen zu erkennen. In Läufen mit vielen kurzen Reads sind die kürzeren Reads manchmal von geringerer Qualität als der Rest.
Betrachten Sie “Read lengths vs Average read quality plot using dots plot”. Ist Ihnen beim Qscore etwas Ungewöhnliches aufgefallen? Können Sie das erklären?
Es gibt keine Reads unter Q20.
Die Qualifikation für HiFi-Reads lautet:
Führen Sie die Qualitätskontrolle mit FastQCtool auf m64011_190830_220126.Q20.subsample.fastq.gz durch und vergleichen Sie die Ergebnisse!
Bewertung der Qualität mit PycoQC - nur Nanopore
PycoQC (Leger and Leonardi 2019) ist ein Datenvisualisierungs- und Qualitätskontrollwerkzeug für Nanopore-Daten. Im Gegensatz zu FastQC/Nanoplot benötigt es eine spezifische sequencing_summary.txt Datei, die von Oxford Nanopore Basecallern wie Guppy oder dem älteren Albacore Basecaller generiert wird.
Eine der Stärken von PycoQC ist, dass es interaktiv und in hohem Maße anpassbar ist, z. B. können Plots beschnitten werden, man kann hinein- und herauszoomen, Bereiche unterwählen und Zahlen exportieren.
Praktische Übung: Qualitätsprüfung von Nanopore-Reads
Erstellen Sie einen neuen Verlauf für diesen Teil und geben Sie ihm einen passenden Namen
Import der Nanopore-Reads nanopore_basecalled-guppy.fastq.gz und sequencing_summary.txt von Zenodo
~270k Reads insgesamt (siehe Basecall-Zusammenfassungstabelle “Alle Reads”)
Bei den meisten Basecalling-Profilen stuft Guppy die Reads als “Pass” ein, wenn der Qscore der Reads mindestens gleich 7 ist.
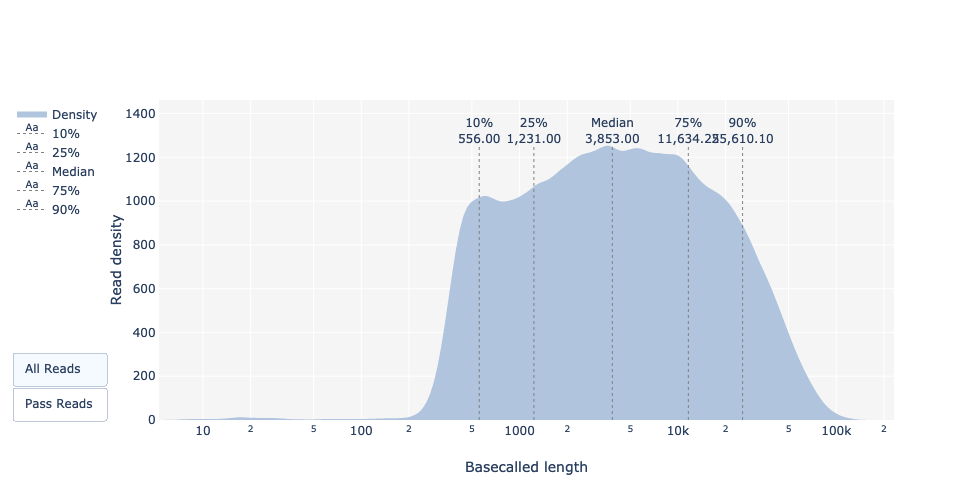
Was ist der Median, das Minimum und das Maximum der Leselänge, was ist der N50?
Die mediane Leselänge und der N50-Wert können für alle sowie für alle bestandenen Reads, d. h. für Reads, die die Guppy-Qualitätseinstellungen (Qscore >= 7) bestanden haben, in der Basecall-Zusammenfassungstabelle gefunden werden.
Für die minimale (195bp) und maximale (256kbp) Leselänge kann sie mit dem Leselängenplot ermittelt werden.
Länge der basisabgerufenen Reads
Wie bei FastQC und Nanoplot zeigt diese Darstellung die Verteilung der Fragmentgrößen in der analysierten Datei. Wie bei PacBio CLR/HiFi haben lange Reads eine variable Länge, und dies zeigt die relativen Mengen der einzelnen Sequenzfragmente unterschiedlicher Größe. In diesem Beispiel ist die Verteilung der Leselänge recht breit gestreut mit einer minimalen Leselänge für die übergebenen Reads um 200bp und einer maximalen Länge von ~150.000bp.
Diese Grafik zeigt die Verteilung der Q-Scores (Q) für jeden Read. Dieser Wert soll eine globale Qualitätsbewertung für jeden Read liefern. Die genaue Definition von Qscores ist: die durchschnittliche Fehlerwahrscheinlichkeit pro Base, ausgedrückt auf der log (Phred) Skala. Bei Nanopore-Daten liegt die Verteilung in der Regel um 10 oder 12 herum. Bei alten Läufen kann die Verteilung niedriger sein, da die Basecalling-Modelle weniger genau sind als neuere Modelle.
Länge der Basecalled Reads vs. Reads PHRED-Qualität
Frage
Wie sehen die mittlere Qualität und die Qualitätsverteilung des Laufs aus?
Die Mehrheit der Reads hat einen Qscore zwischen 8 und 11, was für Nanopore-Daten Standard ist. Beachten Sie, dass für dieselben Daten der verwendete Basecaller (Albacor, Guppy, Bonito), das Modell (fast, hac, sup) und die Toolversion unterschiedliche Ergebnisse liefern können.
Wie bei NanoPlot bietet diese Darstellung eine 2D-Visualisierung des Read-QScores in Abhängigkeit von der Länge.
Abbildung 22: Basecalled reads length vs reads PHRED quality
Ausgabe über die Experimentierzeit
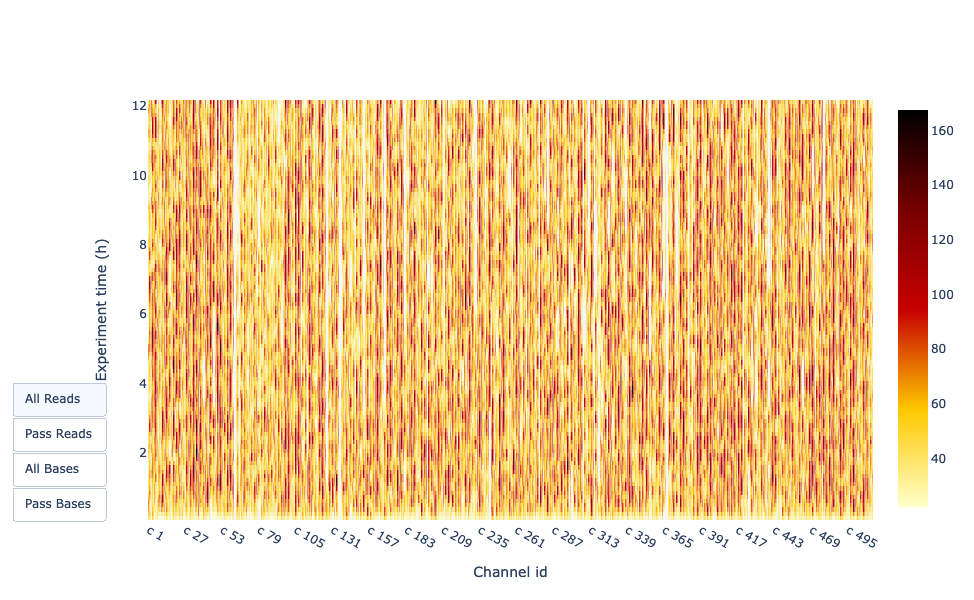
Diese Darstellung gibt Auskunft über die sequenzierten Reads über die Zeit für einen einzelnen Lauf:
Jedes Bild zeigt eine neue Ladung der Fließzelle an (3 + die erste Ladung).
Der Anteil an den gesamten Reads für jedes “Tanken”.
Die Produktion von Reads nimmt mit der Zeit ab:
Der größte Teil des Materials (DNA/RNA) ist sequenziert
Sättigung der Poren
Material-/Porendegradation
…
In diesem Beispiel ist der Beitrag jeder Betankung sehr gering, und es kann als schlechter Lauf betrachtet werden. Der “Cummulative”-Plotbereich (hellblau) zeigt an, dass 50 % aller Reads und fast 50 % aller Basen in den ersten 5 Stunden des 25-Stunden-Experiments produziert wurden. Obwohl es normal ist, dass die Ausbeute mit der Zeit abnimmt, ist ein derartiger Rückgang kein gutes Zeichen.
In diesem Beispiel nahm die Datenproduktion im Laufe der 12 Stunden nur geringfügig ab, während die kumulativen Daten kontinuierlich zunahmen. Das Fehlen einer abfallenden Kurve am Ende des Laufs deutet darauf hin, dass sich noch biologisches Material auf der Fließzelle befindet. Der Lauf wurde beendet, bevor alles sequenziert war. Es ist ein ausgezeichneter Lauf, der sogar als außergewöhnlich bezeichnet werden kann.
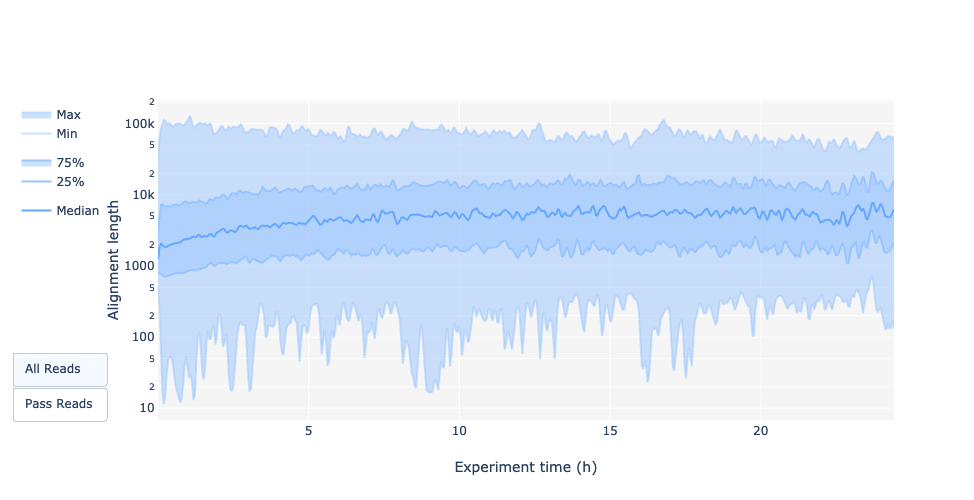
Leselänge über die Experimentierzeit
Frage
Hat sich die Leselänge im Laufe der Zeit verändert? Was könnte der Grund dafür sein?
Im vorliegenden Beispiel nimmt die Leselänge mit der Zeit des Sequenzierungslaufs zu. Eine Erklärung dafür ist, dass die Adapterdichte bei vielen kurzen Fragmenten höher ist und daher die Chance, dass sich ein kürzeres Fragment an eine Pore anlagert, größer ist. Außerdem bewegen sich kürzere Moleküle möglicherweise schneller über den Chip. Mit der Zeit werden die kürzeren Fragmente jedoch seltener, so dass mehr lange Fragmente an den Poren haften und sequenziert werden.
Die Leselänge über die Experimentierzeit sollte stabil sein. Sie kann im Laufe der Zeit leicht ansteigen, da kurze Fragmente zu Beginn übersequenziert werden und im Laufe der Zeit weniger vorhanden sind.
Es gibt einen Überblick über die verfügbaren Poren, die Porenauslastung während des Experiments, die inaktiven Poren und zeigt, ob die Fließzelle gut ausgelastet ist (fast alle Poren werden genutzt). In diesem Fall ist die große Mehrheit der Kanäle/Poren während des gesamten Sequenzierungslaufs inaktiv (weiß), so dass der Lauf als schlecht eingestuft werden kann.
Man hofft auf eine Darstellung, die in der Nähe der X-Achse dunkel ist und bei höheren Y-Werten (mit zunehmender Zeit) nicht zu hell/weiß wird. Je nachdem, ob Sie links “Reads” oder “Basen” auswählen, zeigt die Farbe entweder die Anzahl der Basen oder der Reads pro Zeitintervall an
In diesem Beispiel sind fast alle Poren während des gesamten Laufs aktiv (gelbes/rotes Profil), was auf einen ausgezeichneten Lauf hinweist.
Kommentar: Probier es an!
Führen Sie die Qualitätskontrolle mit FastQCtool und/oder Nanoplottool auf nanopore_basecalled-guppy.fastq.gz durch und vergleichen Sie die Ergebnisse!
Schlussfolgerung
In diesem Lernprogramm haben wir die Qualität der FASTQ-Dateien überprüft, um sicherzustellen, dass die Daten gut aussehen, bevor wir weitere Informationen ableiten. Dieser Schritt ist der übliche erste Schritt für Analysen wie RNA-Seq, ChIP-Seq oder andere OMICs-Analysen, die auf NGS-Daten beruhen. Die Schritte der Qualitätskontrolle sind für jede Art von Sequenzierungsdaten ähnlich:
Qualitätsbewertung mit Tools wie:
Short Reads: FASTQE ( Galaxy version 0.3.1+galaxy0)
Kurz+Lang: FASTQC ( Galaxy version 0.73+galaxy0)
Long Reads: Nanoplot ( Galaxy version 1.41.0+galaxy0)
Nur Nanopore: PycoQC ( Galaxy version 2.5.2+galaxy0)
Trimmen und Filtern nach kurzen Reads mit einem Tool wie Cutadapttool
Du hast das Tutorial abgeschlossen
Please also consider filling out the Feedback Form as well!
Zusammenfassung
Führen Sie vor jeder weiteren bioinformatischen Analyse eine Qualitätskontrolle für jeden Datensatz durch
Beurteilen Sie die Qualitätsmetriken und verbessern Sie die Qualität falls nötig
Überprüfen Sie die Auswirkungen der Qualitätskontrolle
Es stehen verschiedene Werkzeuge zur Verfügung, die zusätzliche Qualitätsmetriken liefern
Bei Paar‑End‑Reads analysieren Sie die Vorwärts‑ und Rückwärtsreads gemeinsam
Häufig gestellte Fragen
Fragen zu diesem Tutorial? Schau auf die verfügbaren FAQ-Seiten und Support-Kanäle
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Ewels, P., M. Magnusson, S. Lundin, and M. K\~ A\textcurrencyller, 2016 MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048. 10.1093/bioinformatics/btw354
De Coster, W., S. D’Hert, D. T. Schultz, M. Cruts, and C. Van Broeckhoven, 2018 NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34: 2666–2669. 10.1093/bioinformatics/bty149
Leger, A., and T. Leonardi, 2019 pycoQC, interactive quality control for Oxford Nanopore Sequencing . Journal of Open Source Software 4: 1236. 10.21105/joss.01236
Jacques, R. M. S., W. M. Maza, S. D. Robertson, A. Lonsdale, C. S. Murray et al., 2021 A Fun Introductory Command Line Lesson: Next Generation Sequencing Quality Analysis with Emoji! CourseSource 8: 10.24918/cs.2021.17
Feedback
Feedback Möglichkeit für Lehrende: Wie lief dein Kurs.
Für Kursteilnehmer/-innen oder Studierende: Über Rückmeldung durch das untenstehende Formular würden wir uns freuen.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{sequence-analysis-quality-control,
author = "Bérénice Batut and Maria Doyle and Alexandre Cormier and Anthony Bretaudeau and Laura Leroi and Erwan Corre and Stéphanie Robin and Cameron Hyde",
title = "Qualitätskontrolle (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/quality-control/tutorial_DE.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Förderung
Diese Personen oder Organisationen unterstützten die Entwicklung dieser Quelle finanziell.
Fragen:

 Open image in new tab
Open image in new tab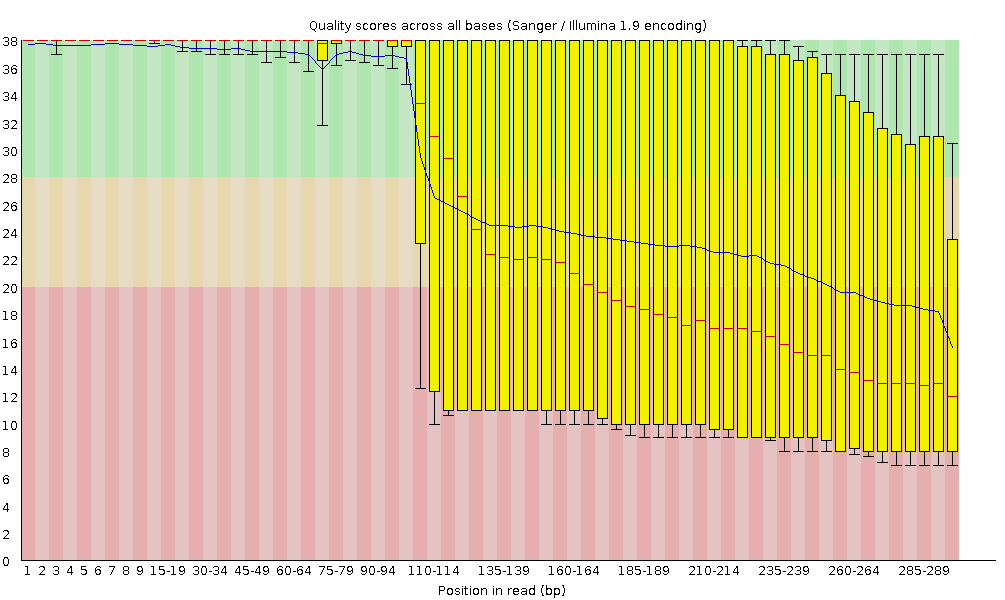 Open image in new tab
Open image in new tab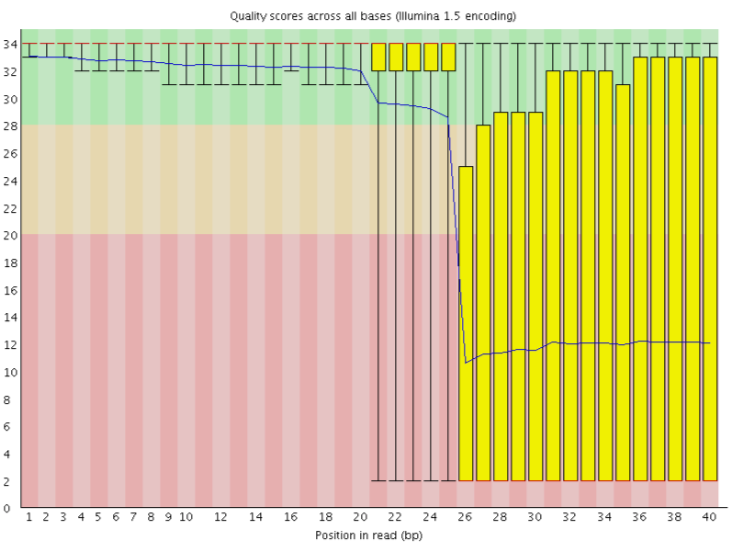
 Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab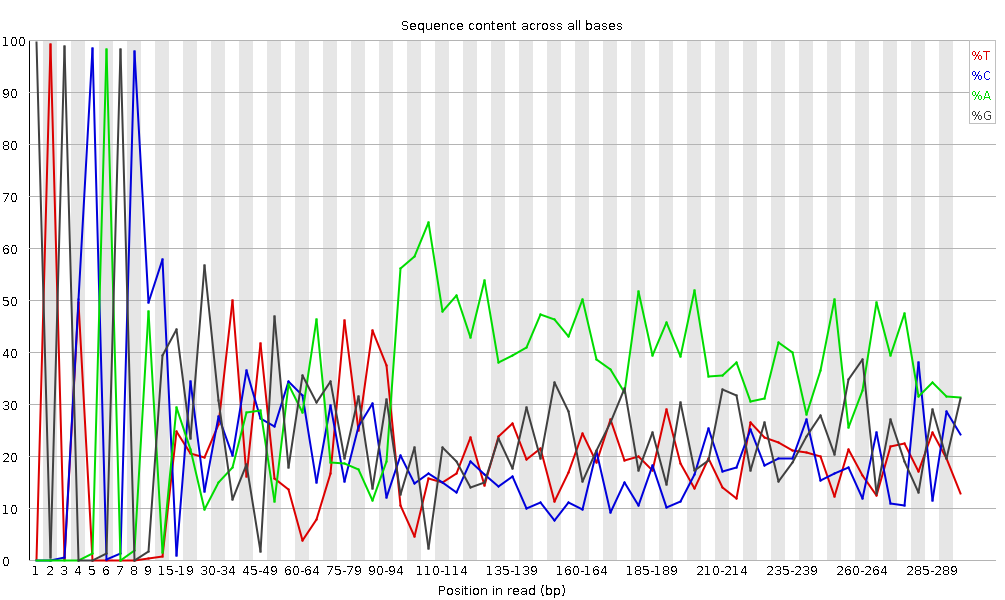 Open image in new tab
Open image in new tab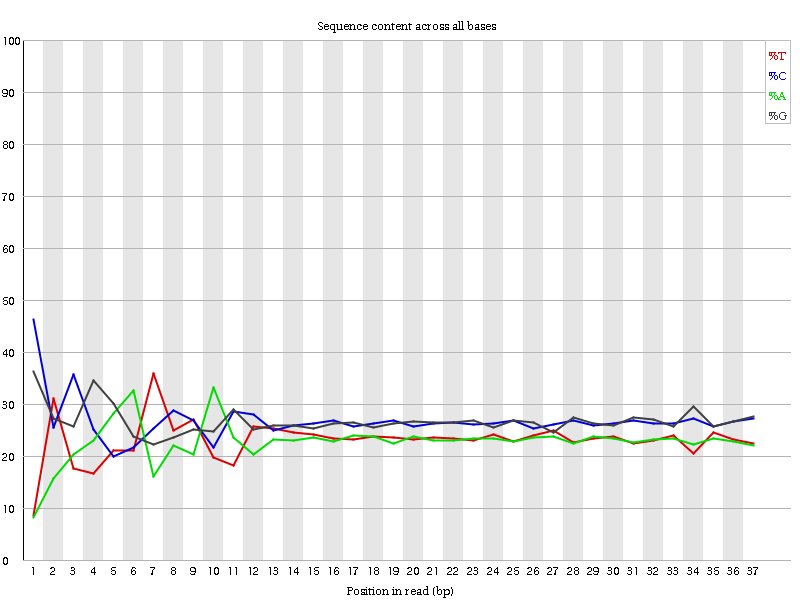
 Open image in new tab
Open image in new tab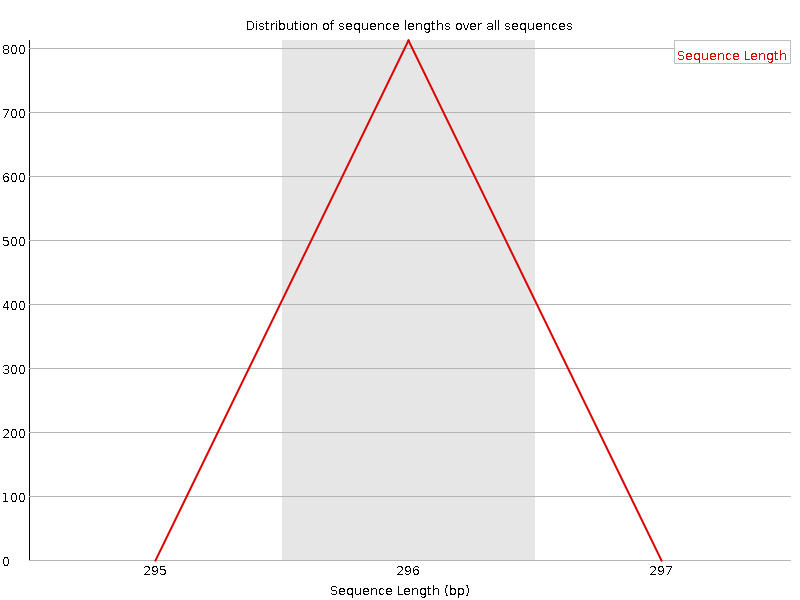 Open image in new tab
Open image in new tab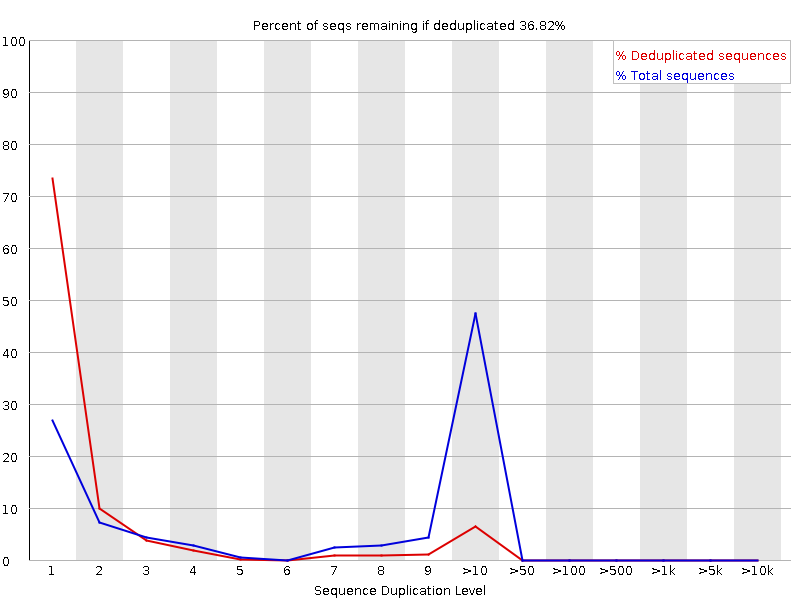 Open image in new tab
Open image in new tab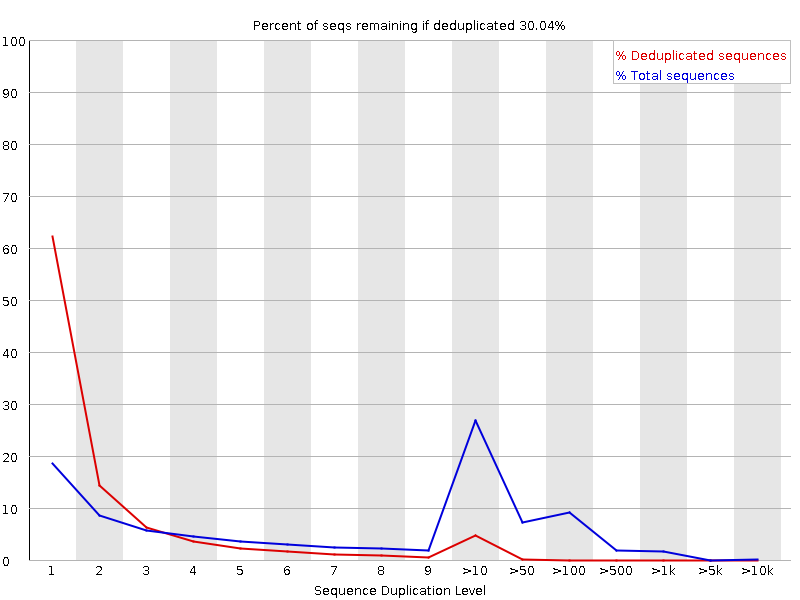
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Wir haben einige rote Streifen, da wir diese Regionen aus den Reads herausgeschnitten haben.
Wir haben jetzt einen Peak hoher Qualität anstelle eines hohen und eines niedrigeren, wie wir ihn vorher hatten.
Da es sich um Amplikon-Daten handelt, sind die Basen nicht mehr gleichmäßig vertreten.
Wir haben jetzt einen einzigen Haupt-GC-Peak aufgrund der Entfernung des Adapters.
Dies ist dasselbe wie vorher, da wir keine Ns in diesen Reads haben.
Wir haben jetzt mehrere Peaks und eine Reihe von Längen, anstelle des einzelnen Peaks, den wir vor dem Trimmen hatten, als alle Sequenzen die gleiche Länge hatten.
Open image in new tab
 Open image in new tab
Open image in new tab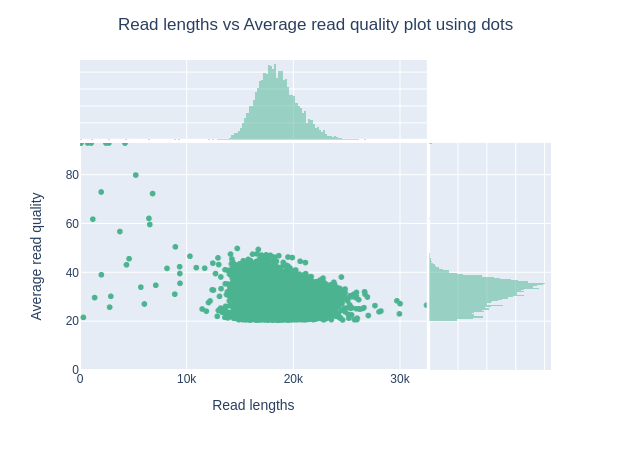 Open image in new tab
Open image in new tabOpen image in new tab
 Open image in new tab
Open image in new tab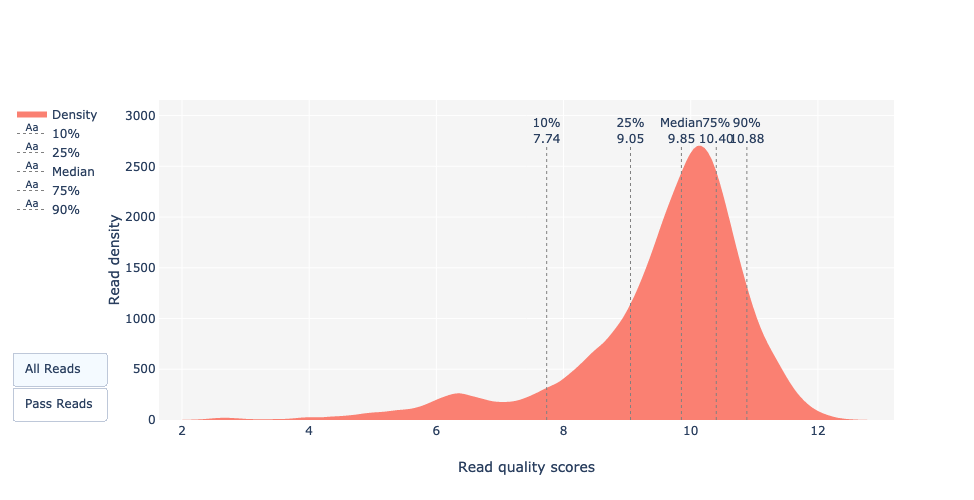 Open image in new tab
Open image in new tab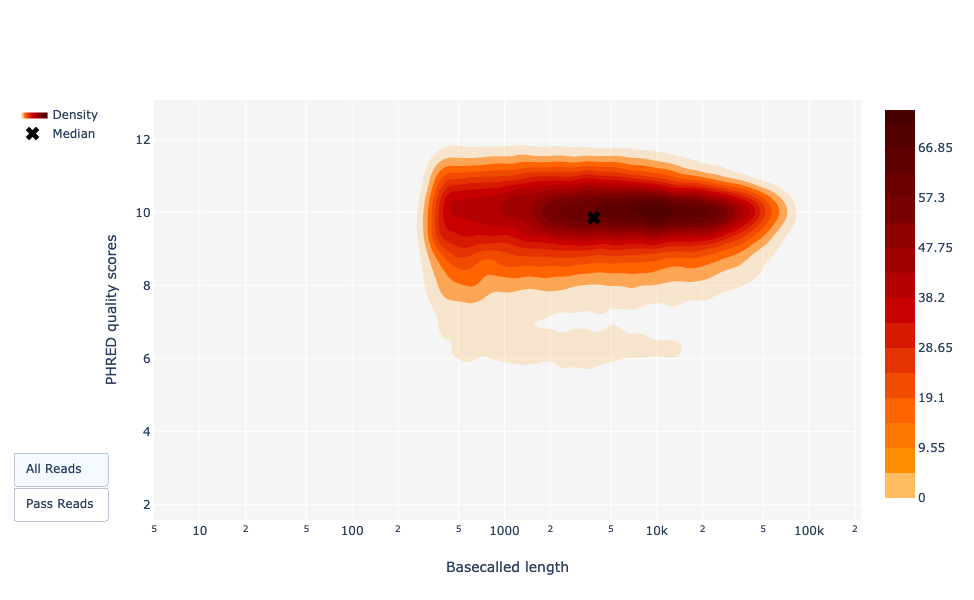 Open image in new tab
Open image in new tab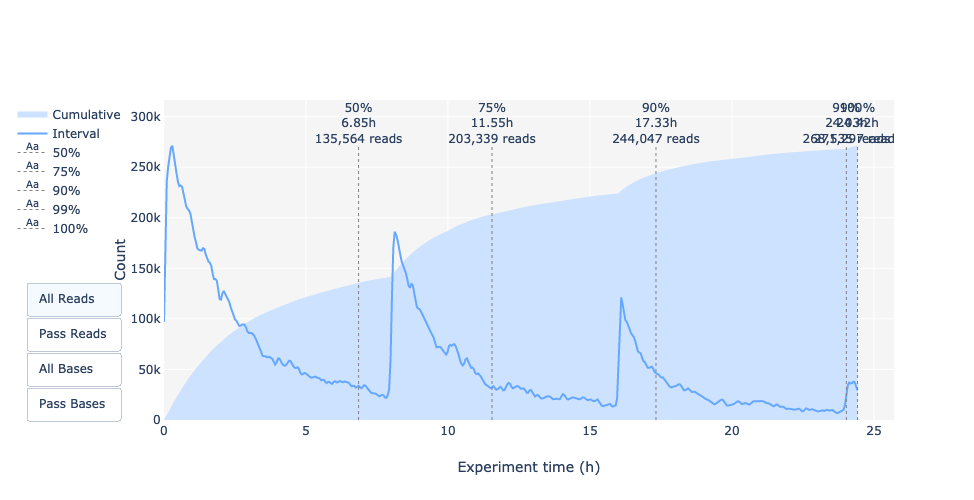 Open image in new tab
Open image in new tab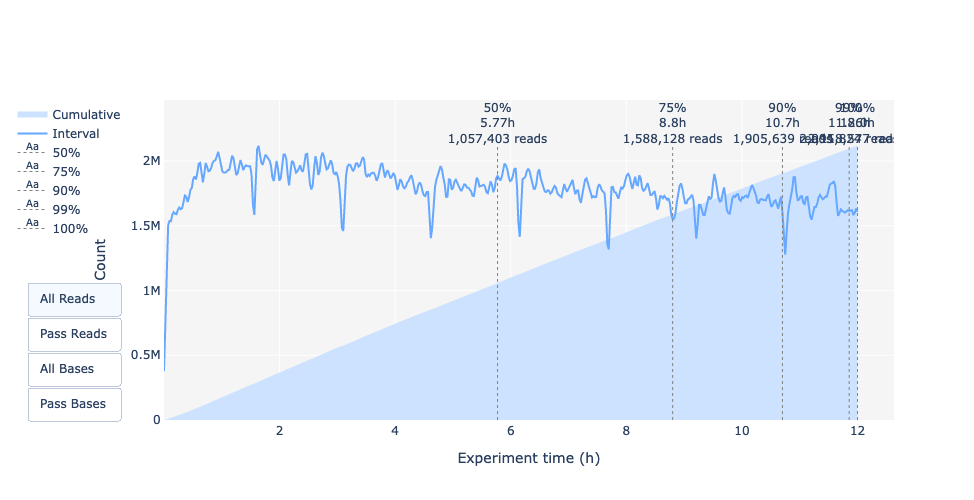
 Open image in new tab
Open image in new tab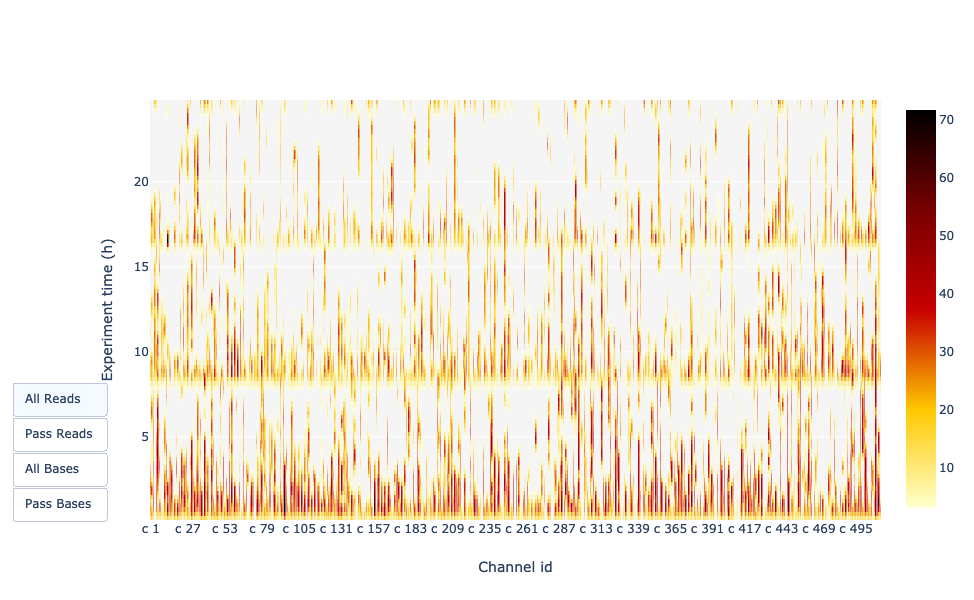 Open image in new tab
Open image in new tab