name: inverse layout: true class: center, middle, inverse <div class="my-header"><span> <a href="/training-material/topics/transcriptomics" title="Return to topic page" ><i class="fa fa-level-up" aria-hidden="true"></i></a> <a href="https://github.com/galaxyproject/training-material/edit/main/topics/transcriptomics/tutorials/rna-seq-viz-with-cummerbund/slides.html"><i class="fa fa-pencil" aria-hidden="true"></i></a> </span></div> <div class="my-footer"><span> <img src="/training-material/assets/images/GTN-60px.png" alt="Galaxy Training Network" style="height: 40px;"/> </span></div> --- <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" class="cover-logo"/> <br/> <br/> # Visualization of RNA-Seq results with CummeRbund <br/> <br/> <div markdown="0"> <div class="contributors-line"> <ul class="text-list"> <li> <a href="/training-material/hall-of-fame/bagnacan/" class="contributor-badge contributor-bagnacan"><img src="https://avatars.githubusercontent.com/bagnacan?s=36" alt="Andrea Bagnacani avatar" width="36" class="avatar" /> Andrea Bagnacani</a></li> </ul> </div> </div> <!-- modified date --> <div class="footnote" style="bottom: 8em;"> <i class="far fa-calendar" aria-hidden="true"></i><span class="visually-hidden">last_modification</span> Updated: <i class="fas fa-fingerprint" aria-hidden="true"></i><span class="visually-hidden">purl</span><abbr title="Persistent URL">PURL</abbr>: <a href="https://gxy.io/GTN:S00098">gxy.io/GTN:S00098</a> </div> <!-- other slide formats (video and plain-text) --> <div class="footnote" style="bottom: 5em;"> <i class="fas fa-file-alt" aria-hidden="true"></i><span class="visually-hidden">text-document</span><a href="slides-plain.html"> Plain-text slides</a> | </div> <!-- usage tips --> <div class="footnote" style="bottom: 2em;"> <strong>Tip: </strong>press <kbd>P</kbd> to view the presenter notes | <i class="fa fa-arrows" aria-hidden="true"></i><span class="visually-hidden">arrow-keys</span> Use arrow keys to move between slides </div> ??? Presenter notes contain extra information which might be useful if you intend to use these slides for teaching. Press `P` again to switch presenter notes off Press `C` to create a new window where the same presentation will be displayed. This window is linked to the main window. Changing slides on one will cause the slide to change on the other. Useful when presenting. --- ## Requirements Before diving into this slide deck, we recommend you to have a look at: - [Introduction to Galaxy Analyses](/training-material/topics/introduction) - [Sequence analysis](/training-material/topics/sequence-analysis) - Quality Control: [<i class="fab fa-slideshare" aria-hidden="true"></i><span class="visually-hidden">slides</span> slides](/training-material/topics/sequence-analysis/tutorials/quality-control/slides.html) - [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](/training-material/topics/sequence-analysis/tutorials/quality-control/tutorial.html) - Mapping: [<i class="fab fa-slideshare" aria-hidden="true"></i><span class="visually-hidden">slides</span> slides](/training-material/topics/sequence-analysis/tutorials/mapping/slides.html) - [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](/training-material/topics/sequence-analysis/tutorials/mapping/tutorial.html) --- ### <i class="far fa-question-circle" aria-hidden="true"></i><span class="visually-hidden">question</span> Questions - How are RNA-Seq results stored? - Why are visualization techniques needed? - How to select our desired subjects for differential gene expression analysis? --- ### <i class="fas fa-bullseye" aria-hidden="true"></i><span class="visually-hidden">objectives</span> Objectives - Manage RNA-Seq results - Extract the desired subject for differential gene expression analysis - Visualize information --- # Why visualization? ??? Data alone does not bring any information: to carry information, data needs to be contextualized. Transcriptomic data is no exception, therefore to organize the growing body of knowledge pertaining RNA-Seq experiments and infer valuable insights, data needs to be organized, annotated, and ultimately visualized. --- ### Where is my data coming from?  <small>[*Wang et al, Nat Rev Genet, 2009*](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2949280/)</small> ??? - RNAs are converted into cDNA fragments through RNA fragmentation - sequencing adaptors (in blue) are then added to each cDNA fragment - cDNA sequences are obtained via NGS sequencing - the obtained reads are subsequently aligned against a reference genome or transcriptome, and classified in-silico as exonic reads, junction reads, and poly-A reads - these three types are then used to outline an expression profile for each gene This is the process that will: - reveal new genes and splice variants - help quantifying cell-specific gene expression within the genome under study But once this pipeline is implemented, how are sequence data going to be analysed and managed? --- ### Bioinformatic tools for RNA-Seq analysis Once the RNA-Seq pipeline is implemented, we still need to handle and analyse all data that is generated. This requires: - computer science skills to be handled - mathematical knowledge to be interpreted --- ### Bioinformatic tools for RNA-Seq analysis  <small>[*Trapnell et al, Nat Protoc, 2012*](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3334321/)</small> ??? - first, reads from each condition are mapped to the reference genome using TopHat - the resulting alignment files are given to Cufflinks, which generates a transcriptome assembly for each condition - the two assemblies are then merged together to provides a uniform basis for calculating of gene and transcript expression in each condition - both reads and merged assemblies are fed to CuffDiff, to calculate expression levels via statistical significance test for the observed changes --- ### Bioinformatic tools for RNA-Seq analysis The last step in our RNA-Seq analysis is CuffDiff. Its output comprises multiple files containing the results of the differential expression analysis. - Gene expression levels are reported as <i>tab-separated</i> values: a simple tabular output that can be viewed with any spreadsheet application. Such files contain statistics, gene-related, and transcript-related attributes .image-75[] - Another way to collect all these data is to organize it within a dedicated database for later consultation. CuffDiff can be instructed to do so .image-50[] ??? - CuffDiff provides analyses of differential expression and regulation at the gene and transcript level - its results are reported in a tab separated format - the overall collection of data is difficult to read to obtain a bird's-eye view of the change of expression - the data can be organized in a SQLite database --- ### Bioinformatic tools for RNA-Seq analysis Whatever storage strategy you opted for, i.e. multiple tab-separated-value files or a SQLite database, all data is still retained within text format. We need to have a bird's-eye view of that data, and <i>make sense</i> of it --- # Visualization --- ### CummeRbund [CummeRbund](http://compbio.mit.edu/cummeRbund/) is an R package for visualizing the results of a CuffDiff output. - Manages, integrates, and visualizes all data produced by CuffDiff - Simplifies data exploration - Provides a bird's-eye view of the expression analysis - Helps creating publication-ready plots --- ### CummeRbund CummeRbund needs to be instructed on which data to be visualized: - <i>Extract CuffDiff</i>'s "Transcript differential expression testing" table - <i>Filter</i> the table on the column storing the significance of a differentially expressed gene - <i>Sort</i> all entries on the basis of most significant differentially expressed gene - Identify the most significant differentially expressed gene --- ### CummeRbund Once the most significant differentially expressed gene has been identified, CummeRbund can generate publication-ready plots to highlight... .image-50[] The expression of all isoforms of the single gene with replicate FPKMs --- ### CummeRbund Once the most significant differentially expressed gene has been identified, CummeRbund can generate publication-ready plots to highlight... .image-50[] The expression bar-plot of all isoforms of a gene with replicate FPKMs --- ### CummeRbund ...and many more .image-50[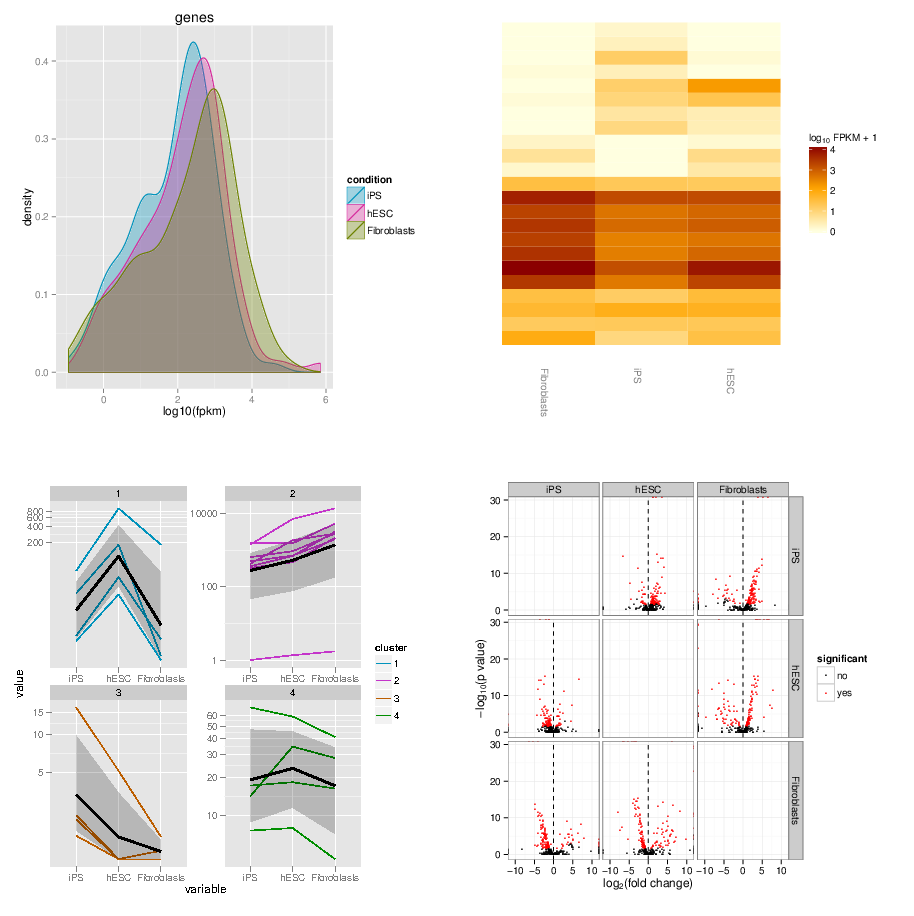] Have a look at [CummerBund's tutorial](https://bioconductor.org/packages/2.11/bioc/vignettes/cummeRbund/inst/doc/cummeRbund-manual.pdf) to overview all possibilities! --- ### <i class="fas fa-key" aria-hidden="true"></i><span class="visually-hidden">keypoints</span> Key points - Extract informations from a SQLite CuffDiff database - Filter and sort results to highlight differential expressed genes of interest - Generate publication-ready visualizations for RNA-Seq analysis results --- ## Thank You! This material is the result of a collaborative work. Thanks to the [Galaxy Training Network](https://training.galaxyproject.org) and all the contributors! <div markdown="0"> <div class="contributors-line"> <table class="contributions"> <tr> <td><abbr title="These people wrote the bulk of the tutorial, they may have done the analysis, built the workflow, and wrote the text themselves.">Author(s)</abbr></td> <td> <a href="/training-material/hall-of-fame/bagnacan/" class="contributor-badge contributor-bagnacan"><img src="https://avatars.githubusercontent.com/bagnacan?s=36" alt="Andrea Bagnacani avatar" width="36" class="avatar" /> Andrea Bagnacani</a> </td> </tr> <tr class="reviewers"> <td><abbr title="These people reviewed this material for accuracy and correctness">Reviewers</abbr></td> <td> <a href="/training-material/hall-of-fame/nsoranzo/" class="contributor-badge contributor-badge-small contributor-nsoranzo"><img src="https://avatars.githubusercontent.com/nsoranzo?s=36" alt="Nicola Soranzo avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/shiltemann/" class="contributor-badge contributor-badge-small contributor-shiltemann"><img src="https://avatars.githubusercontent.com/shiltemann?s=36" alt="Saskia Hiltemann avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/bagnacan/" class="contributor-badge contributor-badge-small contributor-bagnacan"><img src="https://avatars.githubusercontent.com/bagnacan?s=36" alt="Andrea Bagnacani avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/malloryfreeberg/" class="contributor-badge contributor-badge-small contributor-malloryfreeberg"><img src="https://avatars.githubusercontent.com/malloryfreeberg?s=36" alt="Mallory Freeberg avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/njall/" class="contributor-badge contributor-badge-small contributor-njall"><img src="https://avatars.githubusercontent.com/njall?s=36" alt="Niall Beard avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/hexylena/" class="contributor-badge contributor-badge-small contributor-hexylena"><img src="https://avatars.githubusercontent.com/hexylena?s=36" alt="Helena Rasche avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/bebatut/" class="contributor-badge contributor-badge-small contributor-bebatut"><img src="https://avatars.githubusercontent.com/bebatut?s=36" alt="Bérénice Batut avatar" width="36" class="avatar" /></a></td> </tr> </table> </div> </div> <div style="display: flex;flex-direction: row;align-items: center;justify-content: center;"> <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" style="height: 100px;"/> </div> Tutorial Content is licensed under <a rel="license" href="http://creativecommons.org/licenses/by/4.0/">Creative Commons Attribution 4.0 International License</a>.<br/>